

Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation
- PMID: 19255345
- PMCID: PMC2730645
- DOI: 10.1161/CIRCULATIONAHA.108.802918
Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation
Abstract
Background: Although preclinical data suggested that tumor necrosis factor-alpha (TNF) neutralization in heart failure (HF) would be beneficial, clinical trials of TNF antagonists were paradoxically negative. We hypothesized that TNF induces opposing inflammatory and remodeling responses in HF that are TNF-receptor (TNFR) specific.
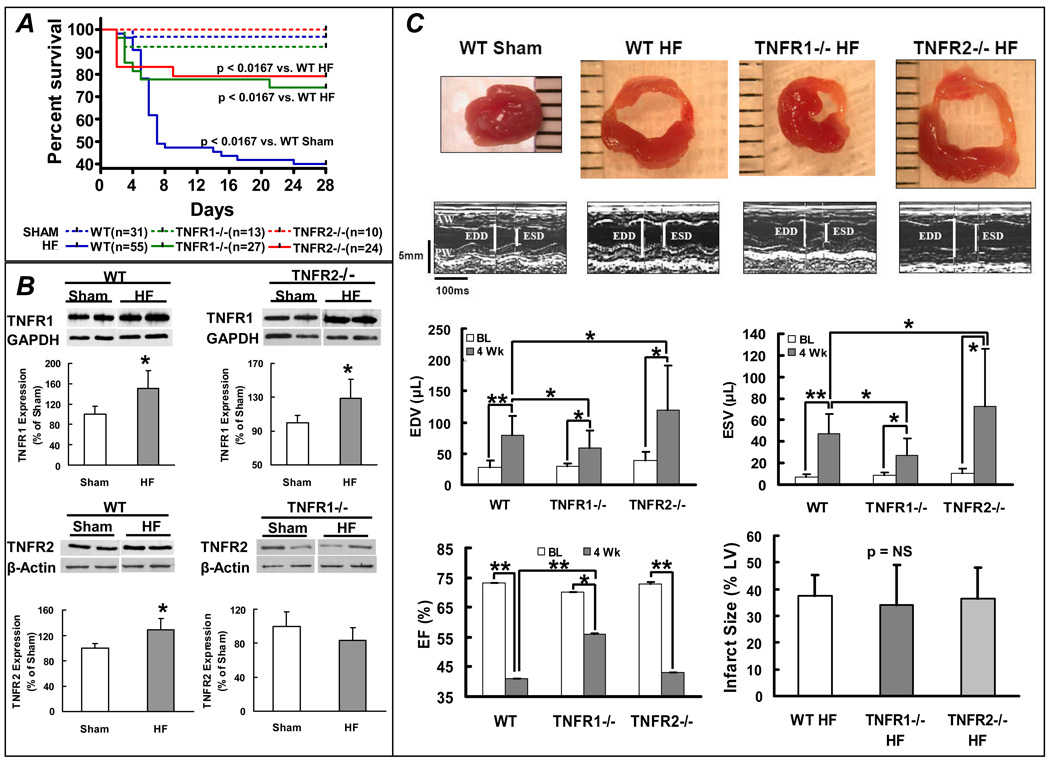
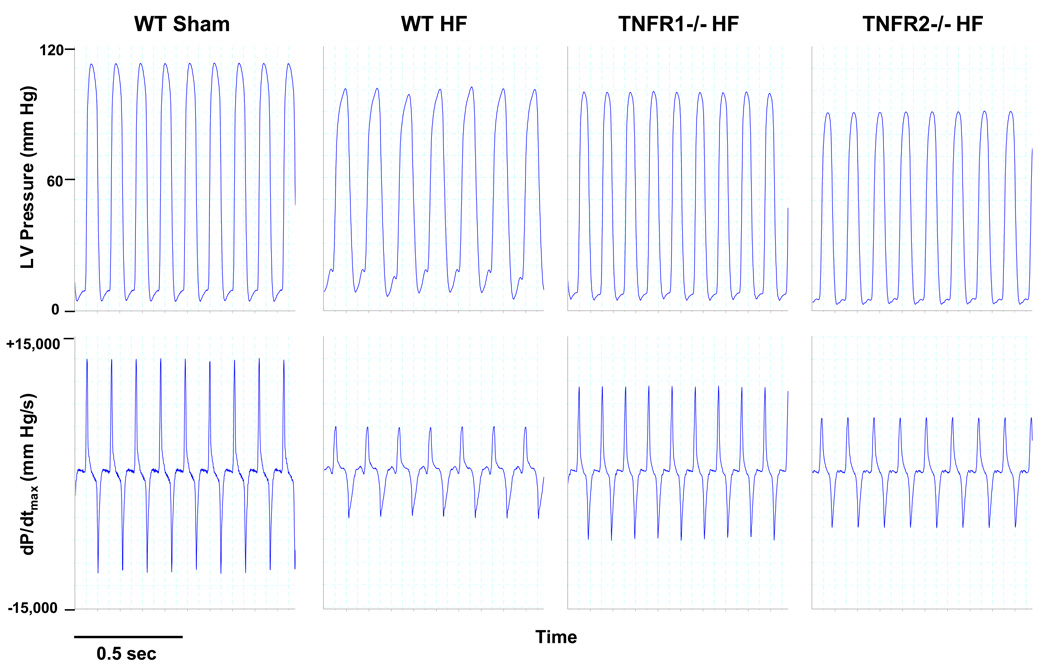
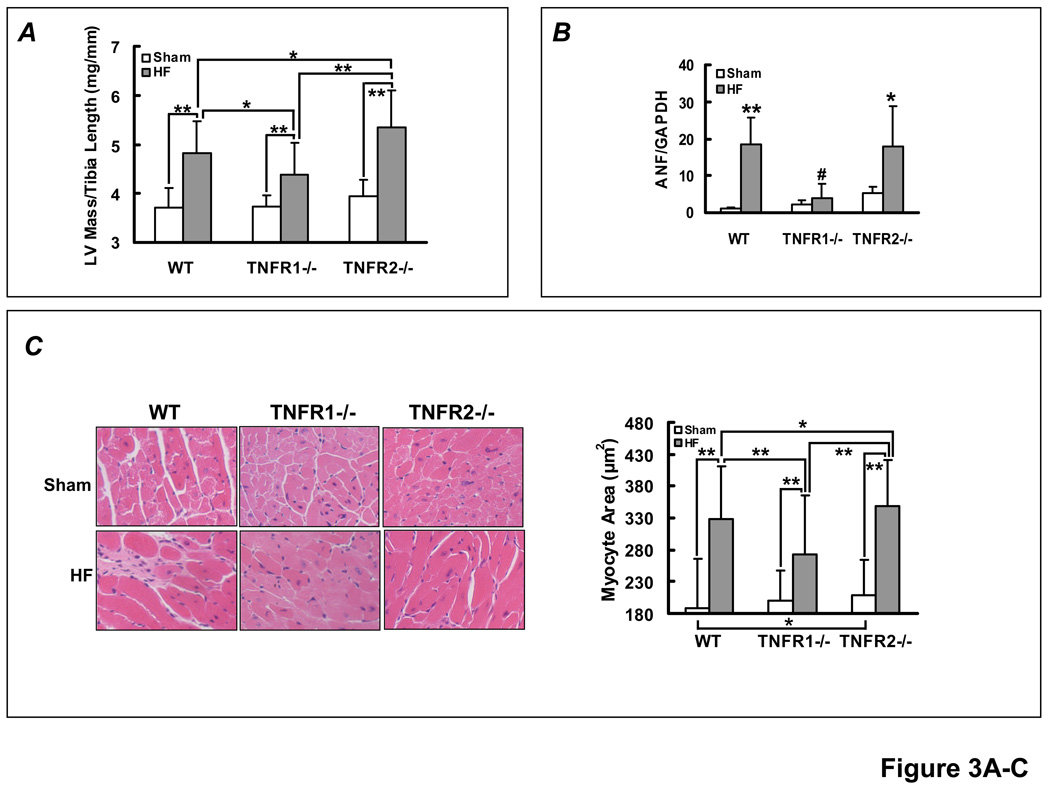
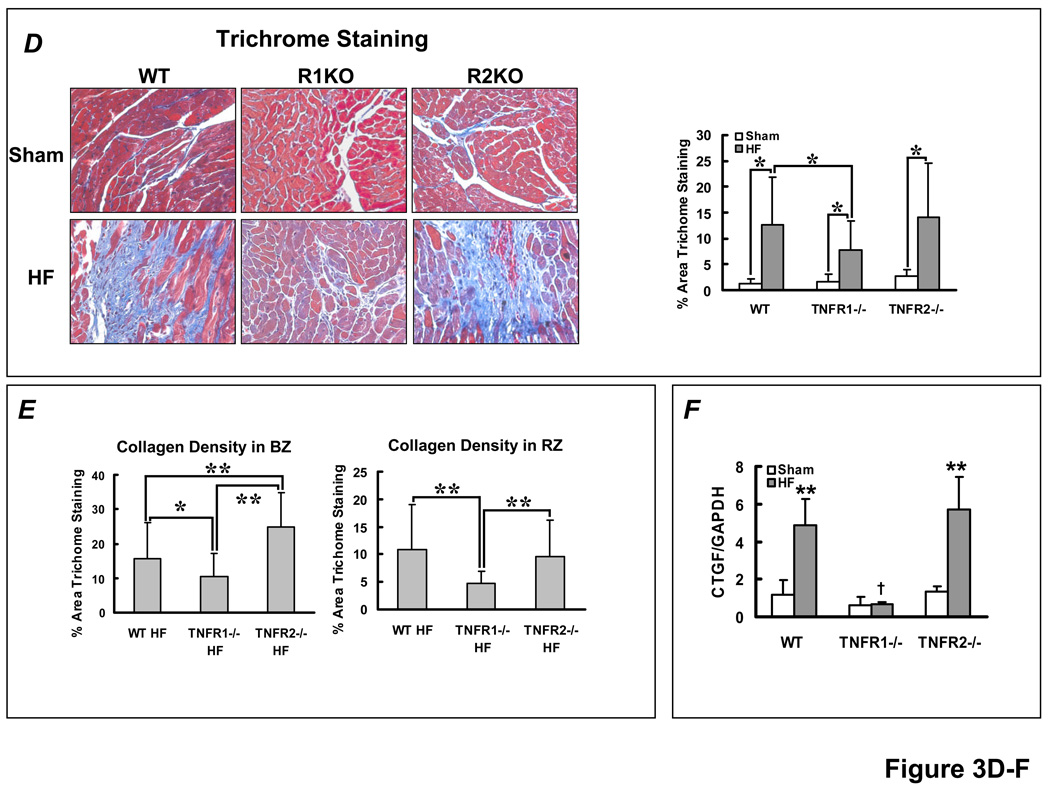
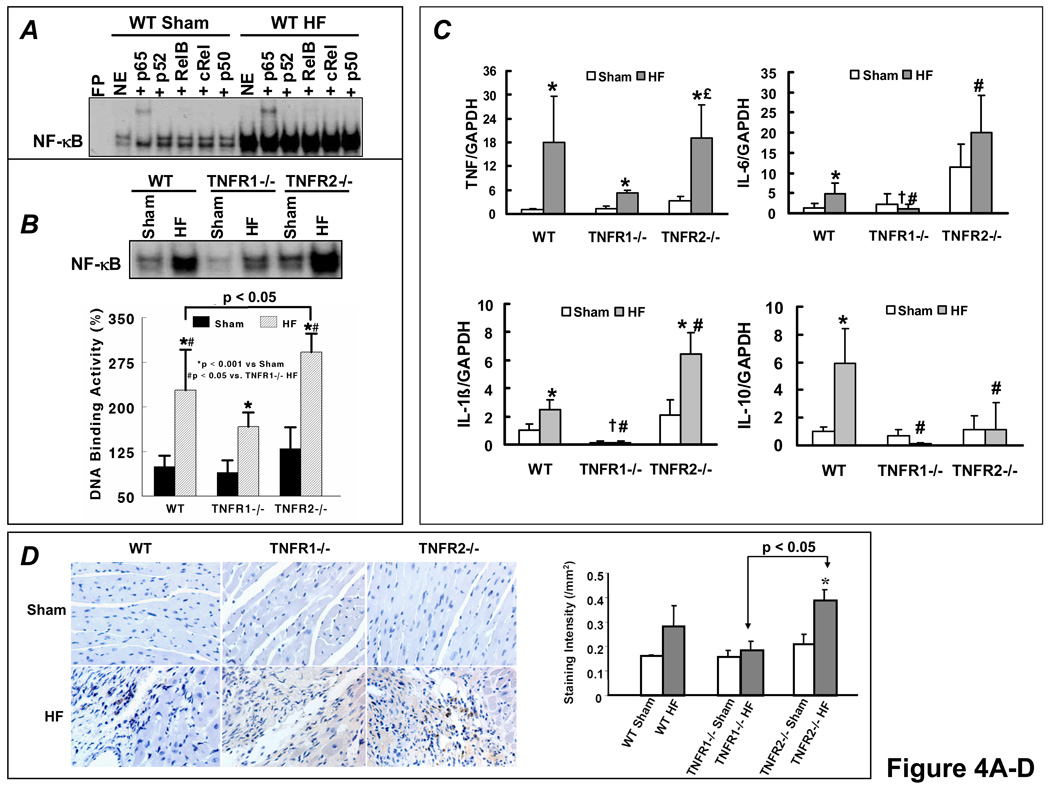
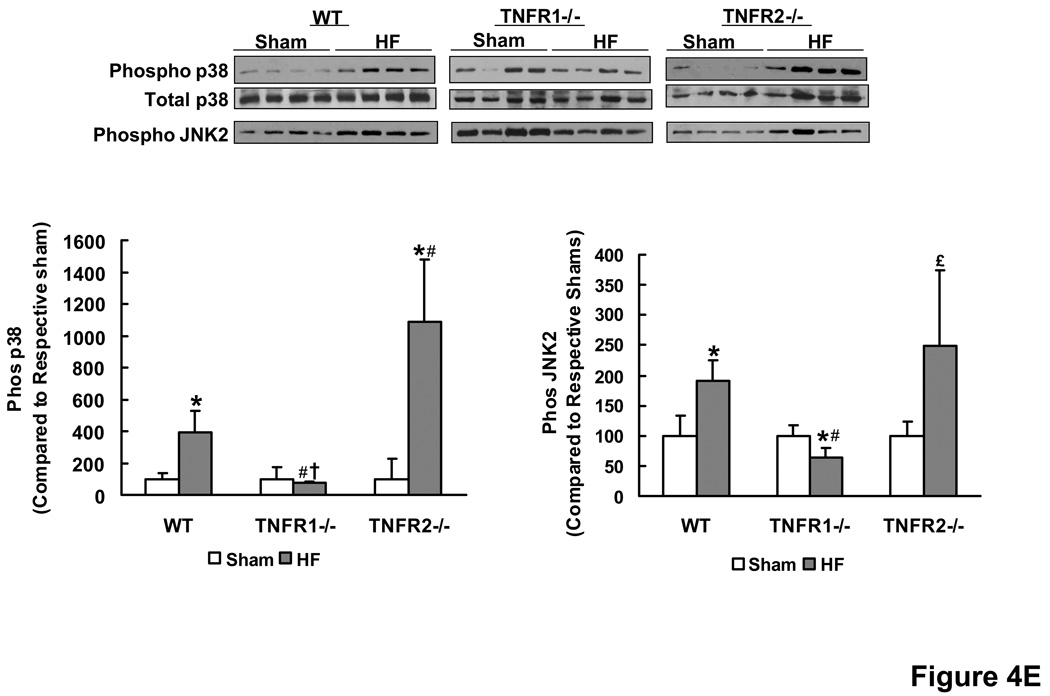
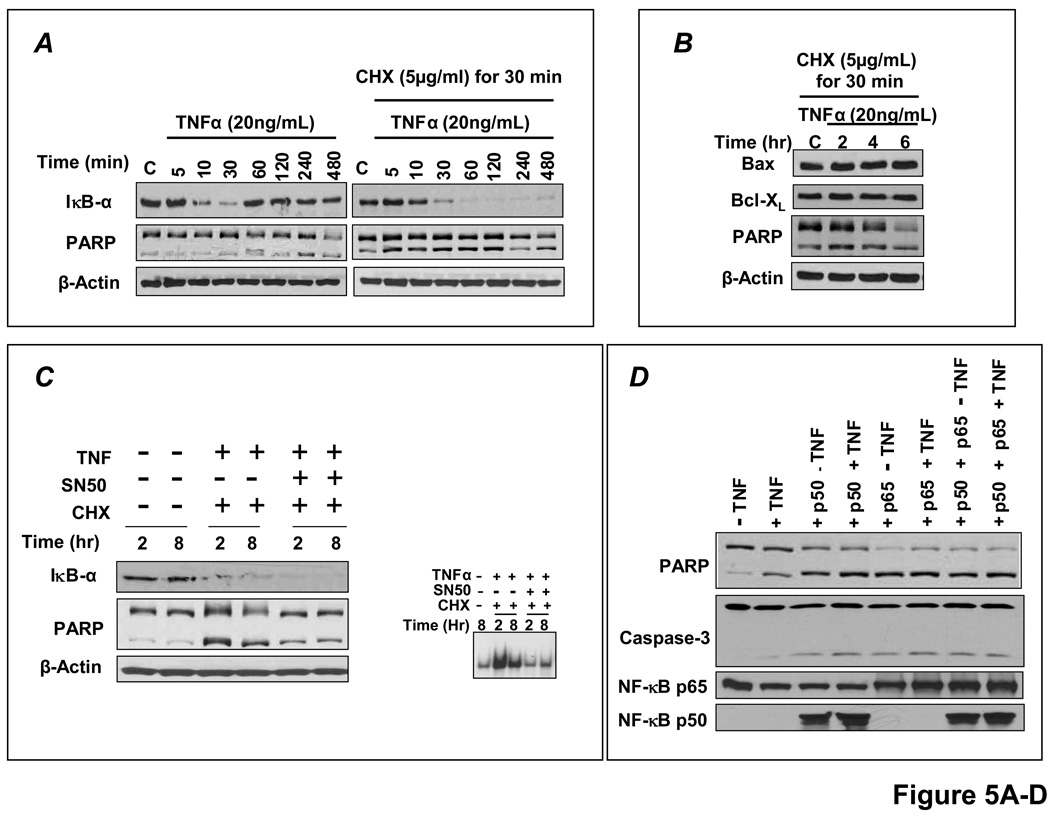
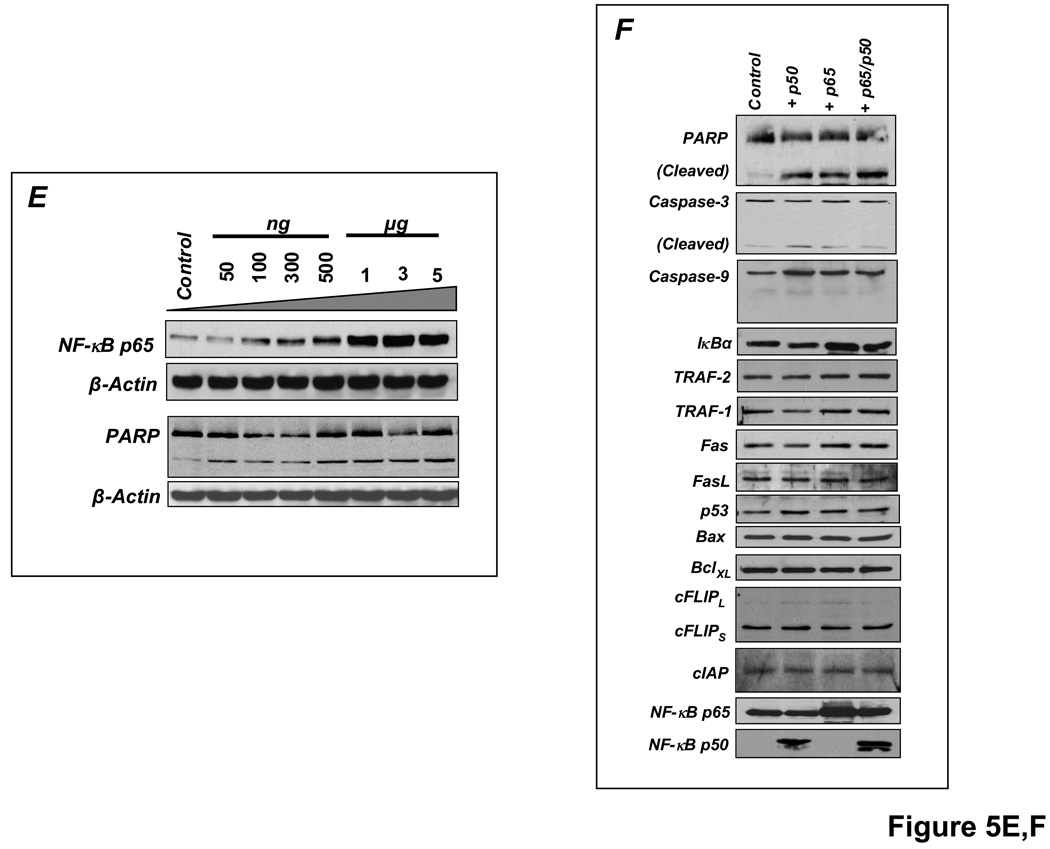
Methods and results: HF was induced in wild-type (WT), TNFR1(-/-), and TNFR2(-/-) mice via coronary ligation. Compared with WT HF, 4-week postinfarction survival was significantly improved in both TNFR1(-/-) and TNFR2(-/-) HF. Compared with sham, WT HF hearts exhibited significant remodeling with robust activation of nuclear factor (NF)-kappaB, p38 mitogen-activated protein kinase, and JNK2 and upregulation of TNF, interleukin (IL)-1beta, IL-6, and IL-10. Compared with WT HF, TNFR1(-/-) HF exhibited (1) improved remodeling, hypertrophy, and contractile function; (2) less apoptosis; and (3) diminished NF-kappaB, p38 mitogen-activated protein kinase, and JNK2 activation and cytokine expression. In contrast, TNFR2(-/-) HF showed exaggerated remodeling and hypertrophy, increased border zone fibrosis, augmented NF-kappaB and p38 mitogen-activated protein kinase activation, higher IL-1beta and IL-6 gene expression, greater activated macrophages, and greater apoptosis. Oxidative stress and diastolic function were improved in both TNFR1(-/-)and TNFR2(-/-) HF. In H9c2 cardiomyocytes, sustained NF-kappaB activation was proapoptotic, an effect dependent on TNFR1 signaling, whereas TNFR2 overexpression attenuated TNF-induced NF-kappaB activation.
Conclusions: TNFR1 and TNFR2 have disparate and opposing effects on remodeling, hypertrophy, NF-kappaB, inflammation, and apoptosis in HF: TNFR1 exacerbates, whereas TNFR2 ameliorates, these events. However, signaling through both receptors is required to induce diastolic dysfunction and oxidative stress. TNFR-specific effects in HF should be considered when therapeutic anti-TNF strategies are developed.
Conflict of interest statement
There are no commercial affiliations or conflicts of interest to disclose.
Figures
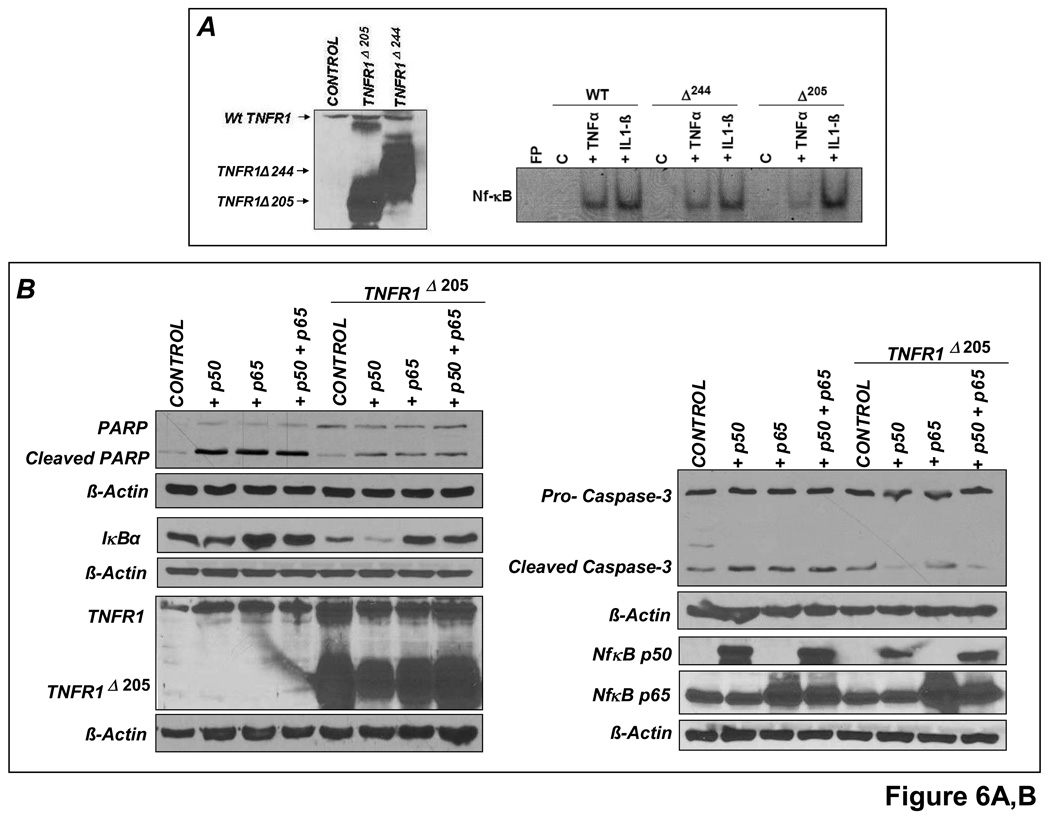
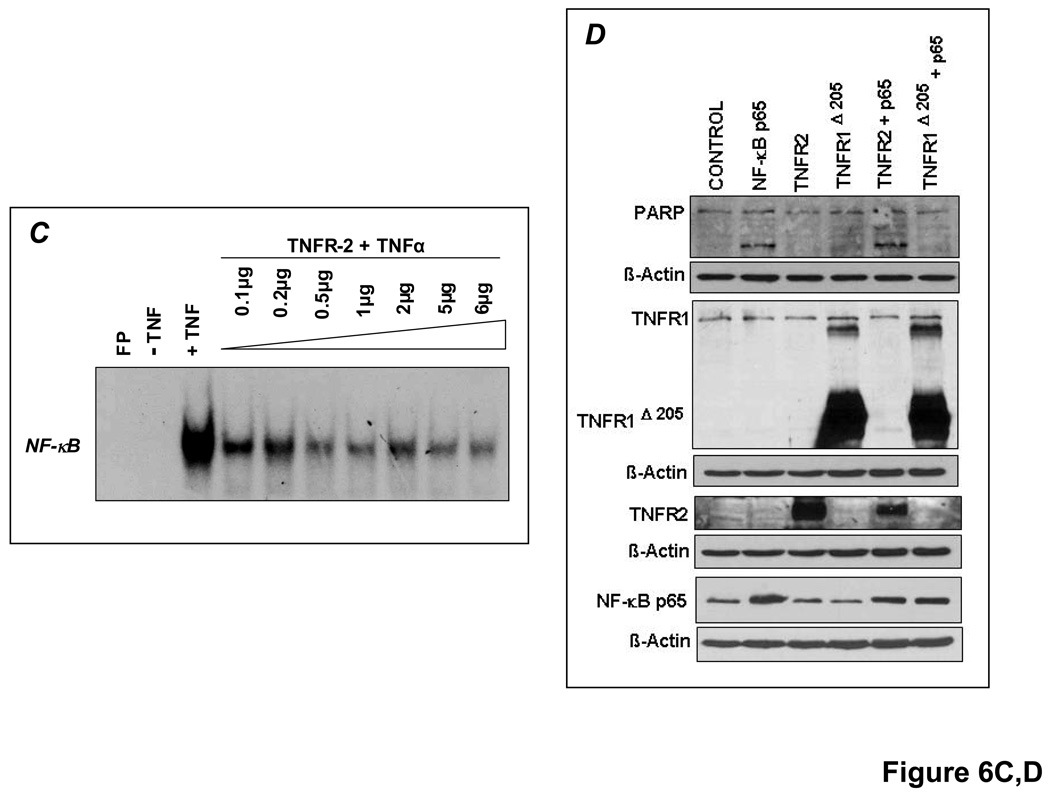
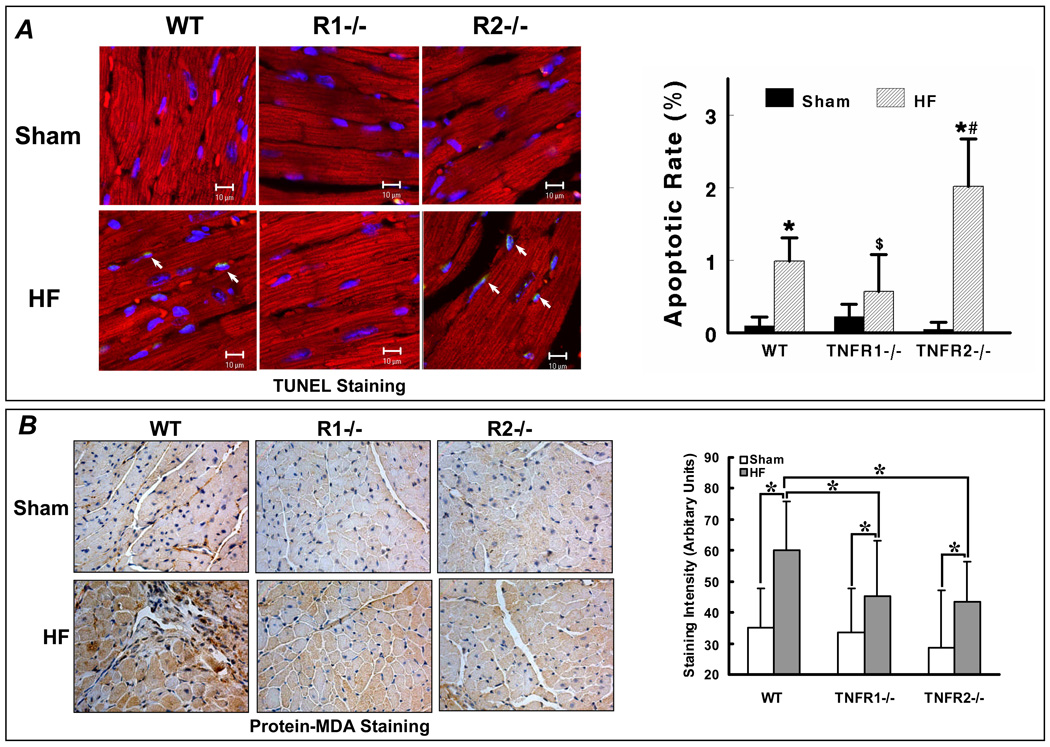
Comment in
-
Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and Yang in myocardial infarction?Circulation. 2009 Mar 17;119(10):1355-7. doi: 10.1161/CIRCULATIONAHA.108.846105. Epub 2009 Mar 2. Circulation. 2009. PMID: 19255338 No abstract available.
References
-
- Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–998. - PubMed
-
- Bozkurt B, Kribbs SB, Clubb FJ, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. - PubMed
-
- Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-α inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287:1813–1820. - PubMed
-
- Iversen PO, Nicolaysen G, Sioud M. DNA enzyme targeting TNF-α mRNA improves hemodynamic performance in rats with postinfarction heart failure. Am J Physiol Heart Circ Physiol. 2001;281:H2211–H2217. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
