

Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity
- PMID: 19256530
- PMCID: PMC2670930
- DOI: 10.1021/tx800464q
Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity
Abstract
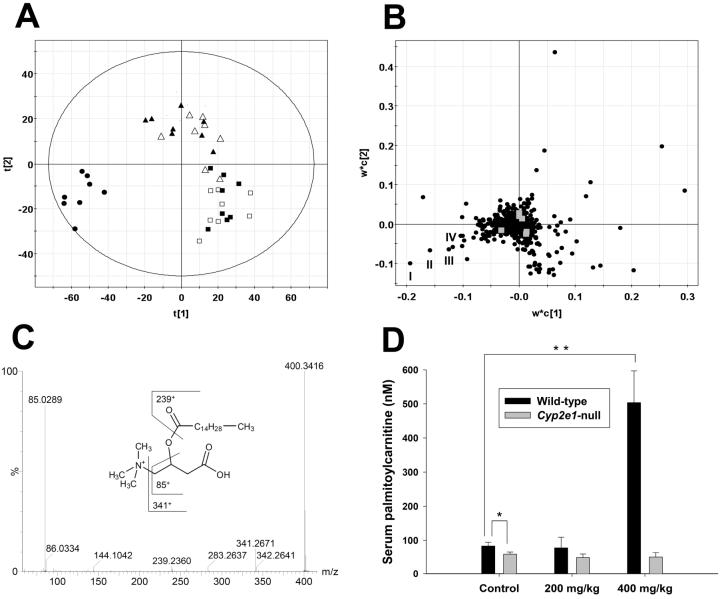
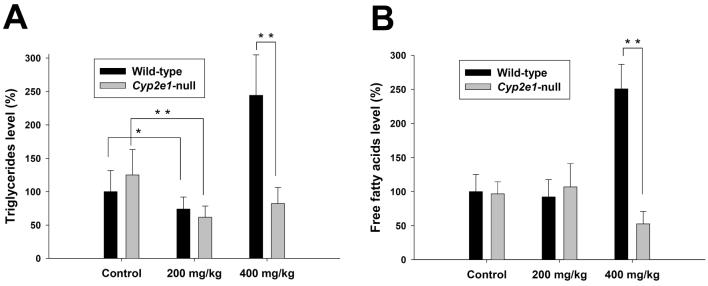
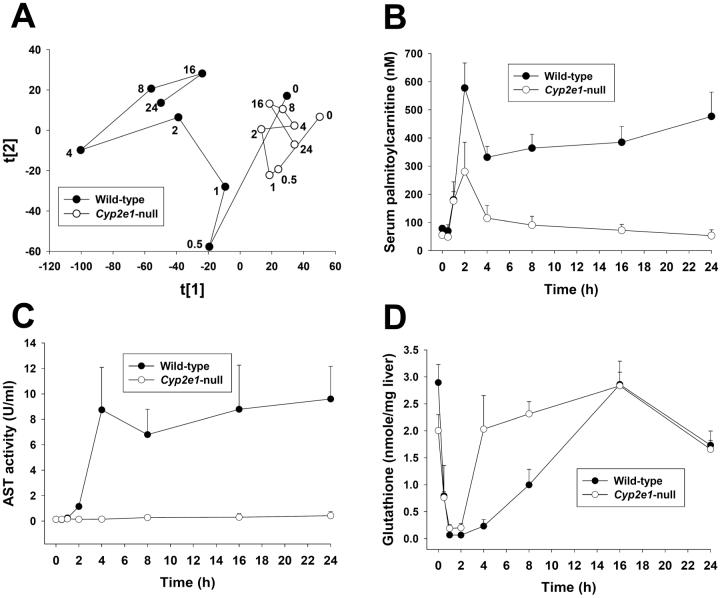
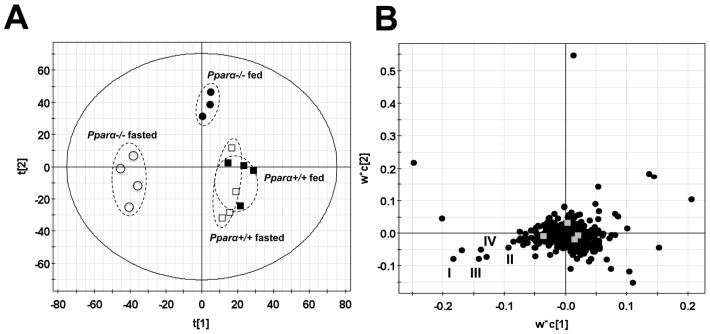
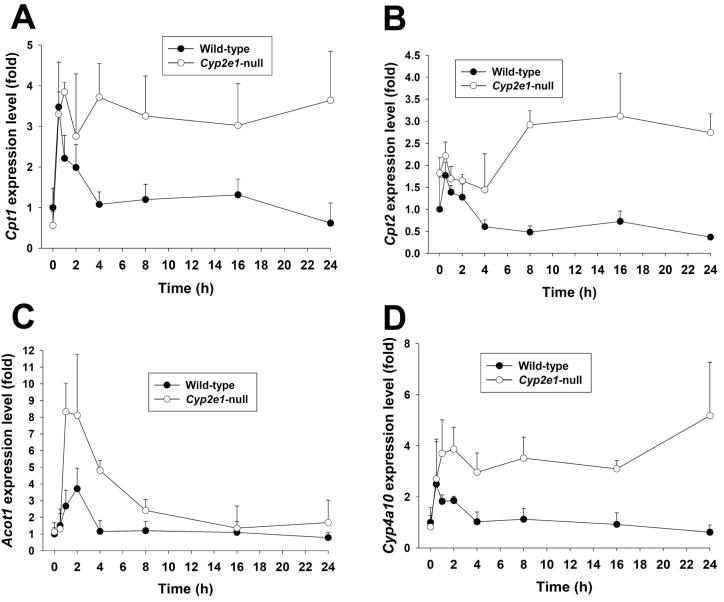
Metabolic bioactivation, glutathione depletion, and covalent binding are the early hallmark events after acetaminophen (APAP) overdose. However, the subsequent metabolic consequences contributing to APAP-induced hepatic necrosis and apoptosis have not been fully elucidated. In this study, serum metabolomes of control and APAP-treated wild-type and Cyp2e1-null mice were examined by liquid chromatography-mass spectrometry (LC-MS) and multivariate data analysis. A dose-response study showed that the accumulation of long-chain acylcarnitines in serum contributes to the separation of wild-type mice undergoing APAP-induced hepatotoxicity from other mouse groups in a multivariate model. This observation, in conjunction with the increase of triglycerides and free fatty acids in the serum of APAP-treated wild-type mice, suggested that APAP treatment can disrupt fatty acid beta-oxidation. A time-course study further indicated that both wild-type and Cyp2e1-null mice had their serum acylcarnitine levels markedly elevated within the early hours of APAP treatment. While remaining high in wild-type mice, serum acylcarnitine levels gradually returned to normal in Cyp2e1-null mice at the end of the 24 h treatment. Distinct from serum aminotransferase activity and hepatic glutathione levels, the pattern of serum acylcarnitine accumulation suggested that acylcarnitines can function as complementary biomarkers for monitoring the APAP-induced hepatotoxicity. An essential role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the regulation of serum acylcarnitine levels was established by comparing the metabolomic responses of wild-type and Ppara-null mice to a fasting challenge. The upregulation of PPARalpha activity following APAP treatment was transient in wild-type mice but was much more prolonged in Cyp2e1-null mice. Overall, serum metabolomics of APAP-induced hepatotoxicity revealed that the CYP2E1-mediated metabolic activation and oxidative stress following APAP treatment can cause irreversible inhibition of fatty acid oxidation, potentially through suppression of PPARalpha-regulated pathways.
Figures
References
-
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, Lee WM. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. - PubMed
-
- Cohen SD, Hoivik DJ, Khairallah EA. Acetaminophen-Induced Hepatotoxicity. In: Plaa GL, Hewitt WR, editors. Toxicology of the Liver. Taylor & Francis; 1998. pp. 159–186.
-
- Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci. 2002;65:135–150. - PubMed
-
- Nicholson JK, Wilson ID. Opinion: understanding `global' systems biology: metabonomics and the continuum of metabolism. Nat Rev Drug Discov. 2003;2:668–676. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
