

Biochemical features and functional implications of the RNA-based T-box regulatory mechanism
- PMID: 19258532
- PMCID: PMC2650889
- DOI: 10.1128/MMBR.00026-08
Biochemical features and functional implications of the RNA-based T-box regulatory mechanism
Abstract
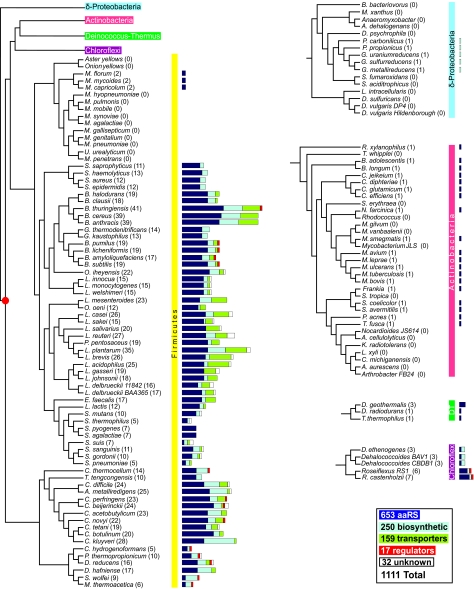
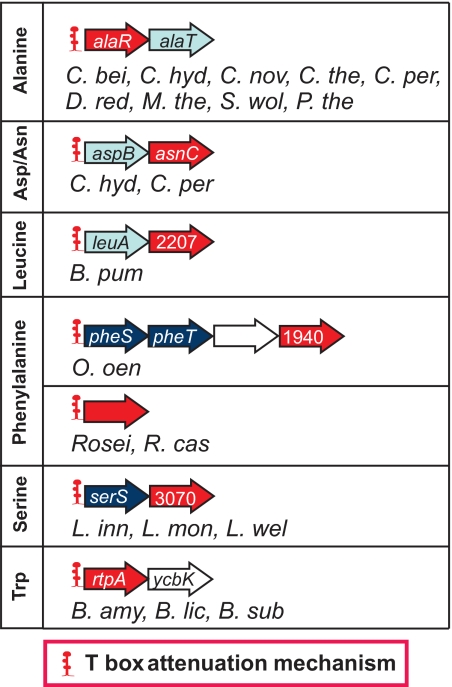
The T-box mechanism is a common regulatory strategy used for modulating the expression of genes of amino acid metabolism-related operons in gram-positive bacteria, especially members of the Firmicutes. T-box regulation is usually based on a transcription attenuation mechanism in which an interaction between a specific uncharged tRNA and the 5' region of the transcript stabilizes an antiterminator structure in preference to a terminator structure, thereby preventing transcription termination. Although single T-box regulatory elements are common, double or triple T-box arrangements are also observed, expanding the regulatory range of these elements. In the present study, we predict the functional implications of T-box regulation in genes encoding aminoacyl-tRNA synthetases, proteins of amino acid biosynthetic pathways, transporters, and regulatory proteins. We also consider the global impact of the use of this regulatory mechanism on cell physiology. Novel biochemical relationships between regulated genes and their corresponding metabolic pathways were revealed. Some of the genes identified, such as the quorum-sensing gene luxS, in members of the Lactobacillaceae were not previously predicted to be regulated by the T-box mechanism. Our analyses also predict an imbalance in tRNA sensing during the regulation of operons containing multiple aminoacyl-tRNA synthetase genes or biosynthetic genes involved in pathways common to more than one amino acid. Based on the distribution of T-box regulatory elements, we propose that this regulatory mechanism originated in a common ancestor of members of the Firmicutes, Chloroflexi, Deinococcus-Thermus group, and Actinobacteria and was transferred into the Deltaproteobacteria by horizontal gene transfer.
Figures








References
-
- Abreu-Goodger, C., N. Ontiveros-Palacios, R. Ciria, and E. Merino. 2004. Conserved regulatory motifs in bacteria: riboswitches and beyond. Trends Genet. 20475-479. - PubMed
-
- Ahn, K. S., and R. G. Wake. 1991. Variations and coding features of the sequence spanning the replication terminus of Bacillus subtilis 168 and W23 chromosomes. Gene 98107-112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
