

Mouse models of Huntington disease: variations on a theme
- PMID: 19259385
- PMCID: PMC2650190
- DOI: 10.1242/dmm.002451
Mouse models of Huntington disease: variations on a theme
Abstract
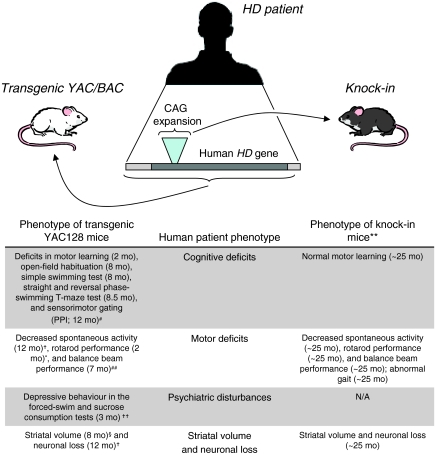
An accepted prerequisite for clinical trials of a compound in humans is the successful alleviation of the disease in animal models. For some diseases, however, successful translation of drug effects from mouse models to the bedside has been limited. One question is whether the current models accurately reproduce the human disease. Here, we examine the mouse models that are available for therapeutic testing in Huntington disease (HD), a late-onset neurodegenerative disorder for which there is no effective treatment. The current mouse models show different degrees of similarity to the human condition. Significant phenotypic differences are seen in mouse models that express either truncated or full-length human, or full-length mouse, mutant huntingtin (mHTT). These differences in phenotypic expression may be attributable to the influences of protein context, mouse strain and a difference in regulatory sequences between the mouse Htt and human HTT genes.
Figures
References
-
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. (1990). Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington’s disease. N. Engl. J. Med. 322, 1293–1298 - PubMed
-
- Bates GP, Valdes J, Hummerich H, Baxendale S, Le Paslier DL, Monaco AP, Tagle D, MacDonald ME, Altherr M, Ross M, et al. (1992). Characterization of a yeast artificial chromosome contig spanning the Huntington’s disease gene candidate region. Nat. Genet. 1, 180–187 - PubMed
-
- Benatar M. (2007). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13 - PubMed
-
- Bessert DA, Gutridge KL, Dunbar JC, Carlock LR. (1995). The identification of a functional nuclear localization signal in the Huntington disease protein. Brain Res. Mol. Brain Res. 33, 165–173 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
