

Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations
- PMID: 19259391
- PMCID: PMC2650198
- DOI: 10.1242/dmm.001263
Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations
Abstract
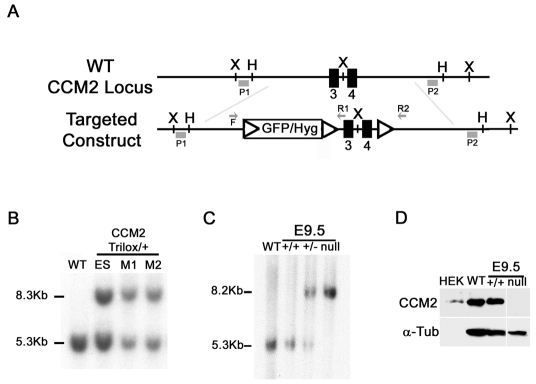
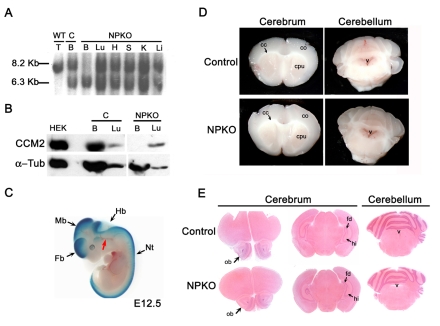
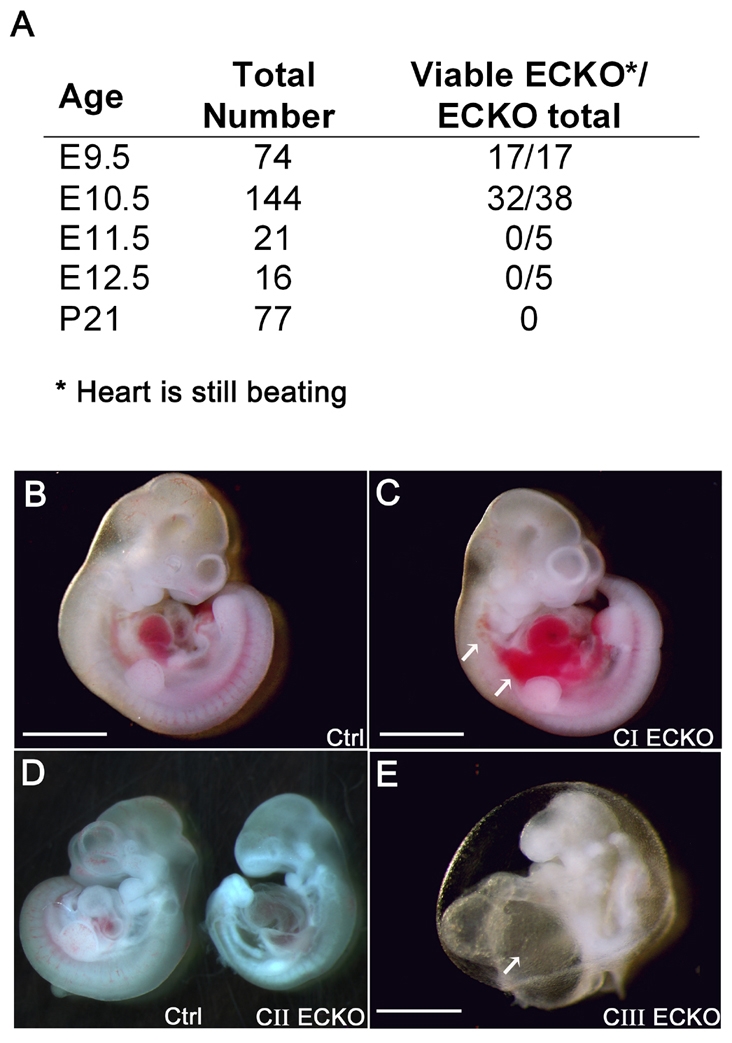
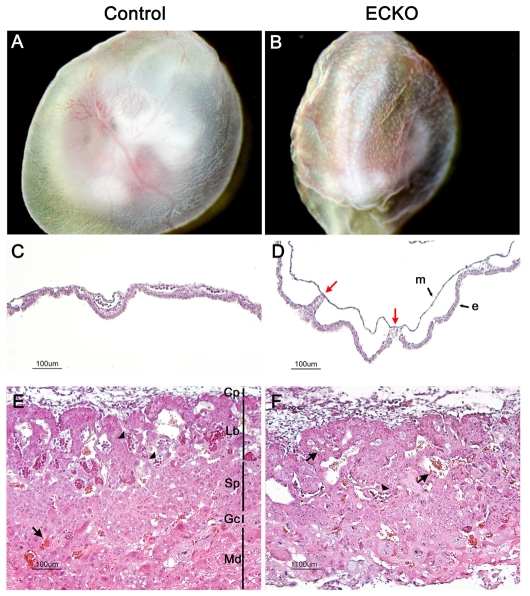
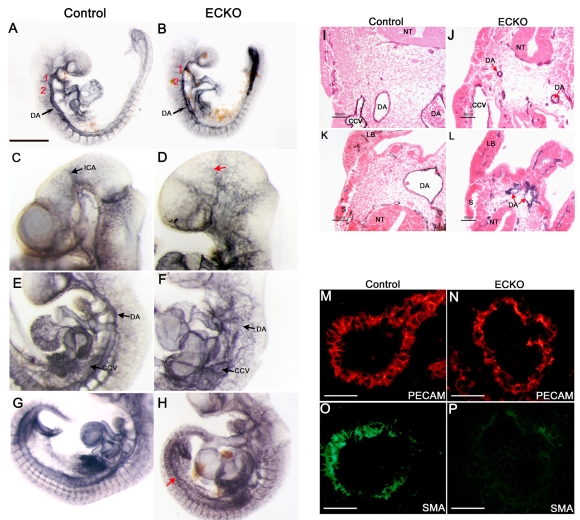
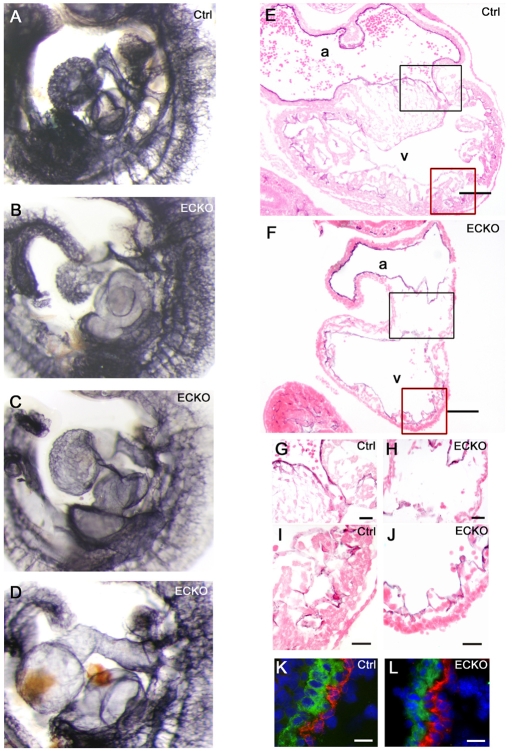
Cerebral cavernous malformations (CCM) are vascular malformations of the brain that lead to cerebral hemorrhages. In 20% of CCM patients, this results from an autosomal dominant condition caused by loss-of-function mutations in one of the three CCM genes. High expression levels of the CCM genes in the neuroepithelium indicate that CCM lesions might be caused by a loss of function of these genes in neural cells rather than in vascular cells. However, their in vivo function, particularly during cerebral angiogenesis, is totally unknown. We developed mice with constitutive and tissue-specific CCM2 deletions to investigate CCM2 function in vivo. Constitutive deletion of CCM2 leads to early embryonic death. Deletion of CCM2 from neuroglial precursor cells does not lead to cerebrovascular defects, whereas CCM2 is required in endothelial cells for proper vascular development. Deletion of CCM2 from endothelial cells severely affects angiogenesis, leading to morphogenic defects in the major arterial and venous blood vessels and in the heart, and results in embryonic lethality at mid-gestation. These findings establish the essential role of endothelial CCM2 for proper vascular development and strongly suggest that the endothelial cell is the primary target in the cascade of events leading from CCM2 mutations to CCM cerebrovascular lesions.
Figures
References
-
- Carmeliet P. (2000). Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
