

GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability
- PMID: 19261619
- PMCID: PMC2670151
- DOI: 10.1074/jbc.M805933200
GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: role in the regulation of tubular paracellular permeability
Abstract
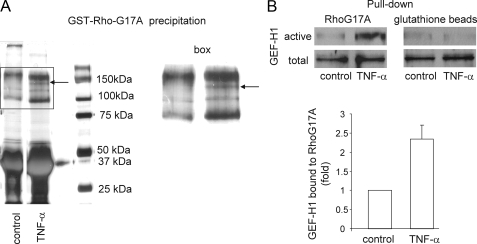
Tumor necrosis factor-alpha (TNF-alpha), an inflammatory cytokine, has been shown to activate the small GTPase Rho, but the underlying signaling mechanisms remained undefined. This general problem is particularly important in the kidney, because TNF-alpha, a major mediator of kidney injury, is known to increase paracellular permeability in tubular epithelia. Here we aimed to determine the effect of TNF-alpha on the Rho pathway in tubular cells (LLC-PK(1) and Madin-Darby canine kidney), define the upstream signaling, and investigate the role of the Rho pathway in the TNF-alpha-induced alterations of paracellular permeability. We show that TNF-alpha induced a rapid and sustained RhoA activation that led to stress fiber formation and Rho kinase-dependent myosin light chain (MLC) phosphorylation. To identify new regulators connecting the TNF receptor to Rho signaling, we applied an affinity precipitation assay with a Rho mutant (RhoG17A), which captures activated GDP-GTP exchange factors (GEFs). Mass spectrometry analysis of the RhoG17A-precipitated proteins identified GEF-H1 as a TNF-alpha-activated Rho GEF. Consistent with a central role of GEF-H1, its down-regulation by small interfering RNA prevented the activation of the Rho pathway. Moreover GEF-H1 and Rho activation are downstream of ERK signaling as the MEK1/2 inhibitor PD98059 mitigated TNF-alpha-induced activation of these proteins. Importantly TNF-alpha enhanced the ERK pathway-dependent phosphorylation of Thr-678 of GEF-H1 that was key for activation. Finally the TNF-alpha-induced paracellular permeability increase was absent in LLC-PK(1) cells stably expressing a non-phosphorylatable, dominant negative MLC. In summary, we have identified the ERK/GEF-H1/Rho/Rho kinase/phospho-MLC pathway as the mechanism mediating TNF-alpha-induced elevation of tubular epithelial permeability, which in turn might contribute to kidney injury.
Figures






Similar articles
-
Extracellular signal-regulated kinase and GEF-H1 mediate depolarization-induced Rho activation and paracellular permeability increase.Am J Physiol Cell Physiol. 2010 Jun;298(6):C1376-87. doi: 10.1152/ajpcell.00408.2009. Epub 2010 Mar 17. Am J Physiol Cell Physiol. 2010. PMID: 20237148 Free PMC article.
-
The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium.J Biol Chem. 2011 Mar 18;286(11):9268-79. doi: 10.1074/jbc.M110.179903. Epub 2011 Jan 6. J Biol Chem. 2011. PMID: 21212278 Free PMC article.
-
Central role of the exchange factor GEF-H1 in TNF-α-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells.Mol Biol Cell. 2013 Apr;24(7):1068-82. doi: 10.1091/mbc.E12-09-0661. Epub 2013 Feb 6. Mol Biol Cell. 2013. PMID: 23389627 Free PMC article.
-
Regulation and functions of the RhoA regulatory guanine nucleotide exchange factor GEF-H1.Small GTPases. 2021 Sep-Nov;12(5-6):358-371. doi: 10.1080/21541248.2020.1840889. Epub 2020 Oct 30. Small GTPases. 2021. PMID: 33126816 Free PMC article. Review.
-
Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis?Trends Cell Biol. 2008 May;18(5):210-9. doi: 10.1016/j.tcb.2008.02.006. Epub 2008 Apr 3. Trends Cell Biol. 2008. PMID: 18394899 Review.
Cited by
-
Stimulation of Rho signaling by pathologic mechanical stretch is a "second hit" to Rho-independent lung injury induced by IL-6.Am J Physiol Lung Cell Mol Physiol. 2012 May 1;302(9):L965-75. doi: 10.1152/ajplung.00292.2011. Epub 2012 Feb 17. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22345573 Free PMC article.
-
Bone resorption by osteoclasts involves fine tuning of RHOA activity by its microtubule-associated exchange factor GEF-H1.Front Physiol. 2024 Jan 19;15:1342024. doi: 10.3389/fphys.2024.1342024. eCollection 2024. Front Physiol. 2024. PMID: 38312316 Free PMC article.
-
Novel role of stathmin in microtubule-dependent control of endothelial permeability.FASEB J. 2012 Sep;26(9):3862-74. doi: 10.1096/fj.12-207746. Epub 2012 Jun 14. FASEB J. 2012. PMID: 22700873 Free PMC article.
-
Claudin-2 suppresses GEF-H1, RHOA, and MRTF, thereby impacting proliferation and profibrotic phenotype of tubular cells.J Biol Chem. 2019 Oct 18;294(42):15446-15465. doi: 10.1074/jbc.RA118.006484. Epub 2019 Sep 3. J Biol Chem. 2019. PMID: 31481470 Free PMC article.
-
IQGAP1 regulates endothelial barrier function via EB1-cortactin cross talk.Mol Cell Biol. 2014 Sep 15;34(18):3546-58. doi: 10.1128/MCB.00248-14. Epub 2014 Jul 14. Mol Cell Biol. 2014. PMID: 25022754 Free PMC article.
References
-
- Baud, V., and Karin, M. (2001) Trends Cell Biol. 11, 372-377 - PubMed
-
- Wajant, H., Pfizenmaier, K., and Scheurich, P. (2003) Cell Death Differ. 10, 45-65 - PubMed
-
- Vielhauer, V., and Mayadas, T. N. (2007) Semin. Nephrol. 27, 286-308 - PubMed
-
- Pascher, A., and Klupp, J. (2005) BioDrugs 19, 211-231 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
