

Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase
- PMID: 19262568
- PMCID: PMC2683695
- DOI: 10.1038/emboj.2009.43
Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase
Abstract
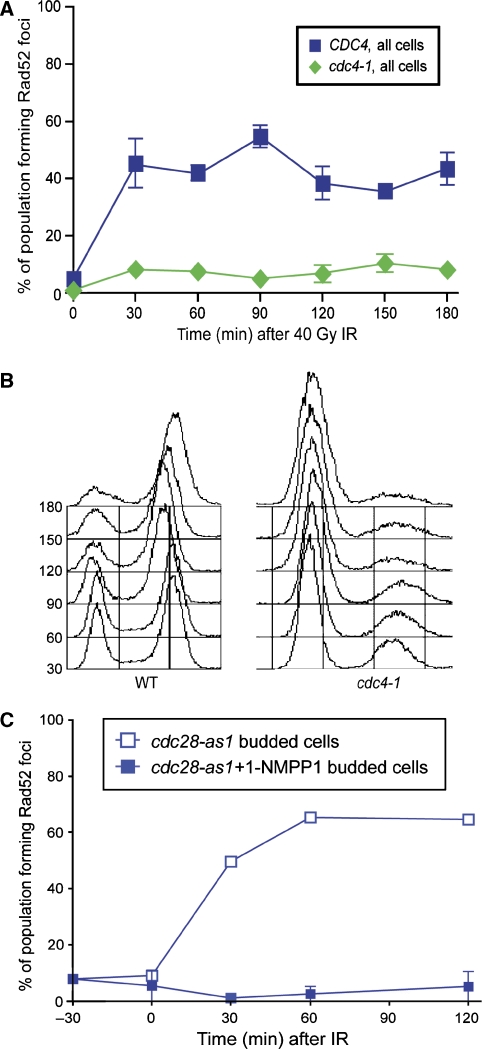
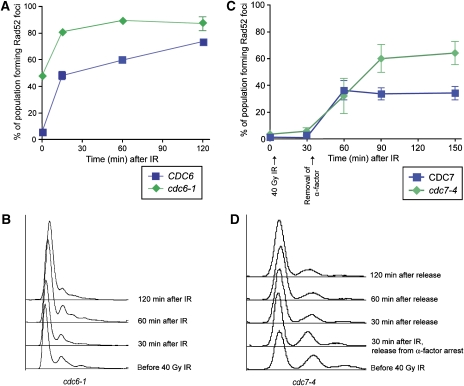
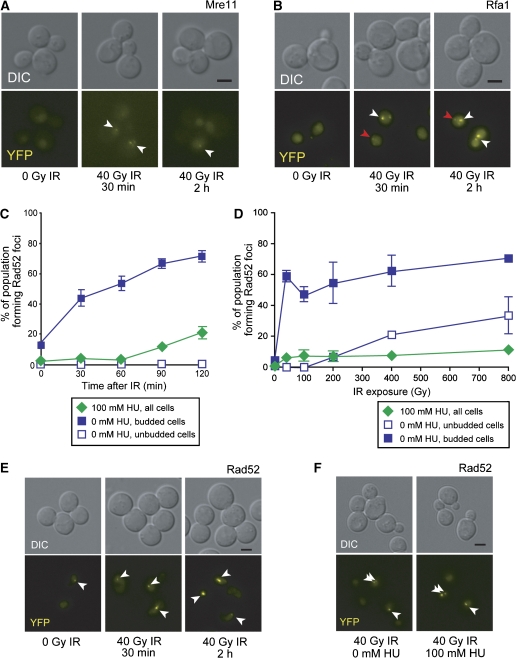
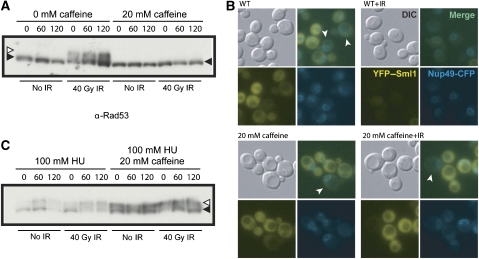
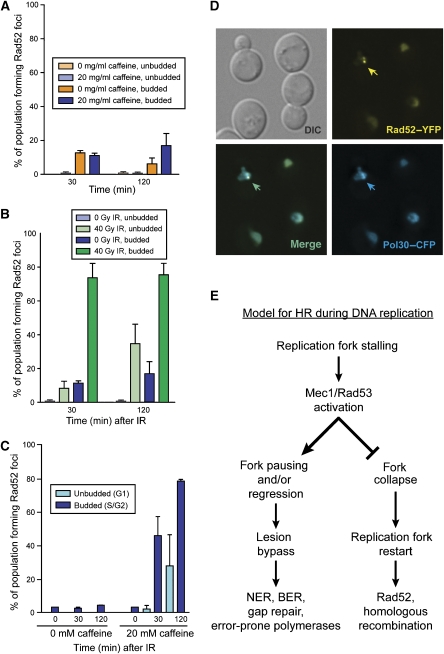
Recruitment of the homologous recombination machinery to sites of double-strand breaks is a cell cycle-regulated event requiring entry into S phase and CDK1 activity. Here, we demonstrate that the central recombination protein, Rad52, forms foci independent of DNA replication, and its recruitment requires B-type cyclin/CDK1 activity. Induction of the intra-S-phase checkpoint by hydroxyurea (HU) inhibits Rad52 focus formation in response to ionizing radiation. This inhibition is dependent upon Mec1/Tel1 kinase activity, as HU-treated cells form Rad52 foci in the presence of the PI3 kinase inhibitor caffeine. These Rad52 foci colocalize with foci formed by the replication clamp PCNA. These results indicate that Mec1 activity inhibits the recruitment of Rad52 to both sites of DNA damage and stalled replication forks during the intra-S-phase checkpoint. We propose that B-type cyclins promote the recruitment of Rad52 to sites of DNA damage, whereas Mec1 inhibits spurious recombination at stalled replication forks.
Figures
References
-
- Bartrand AJ, Iyasu D, Brush GS (2004) DNA stimulates Mec1-mediated phosphorylation of replication protein A. J Biol Chem 279: 26762–26767 - PubMed
-
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407: 395–401 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
