

Oxidative stress induces degradation of mitochondrial DNA
- PMID: 19264794
- PMCID: PMC2677867
- DOI: 10.1093/nar/gkp100
Oxidative stress induces degradation of mitochondrial DNA
Abstract
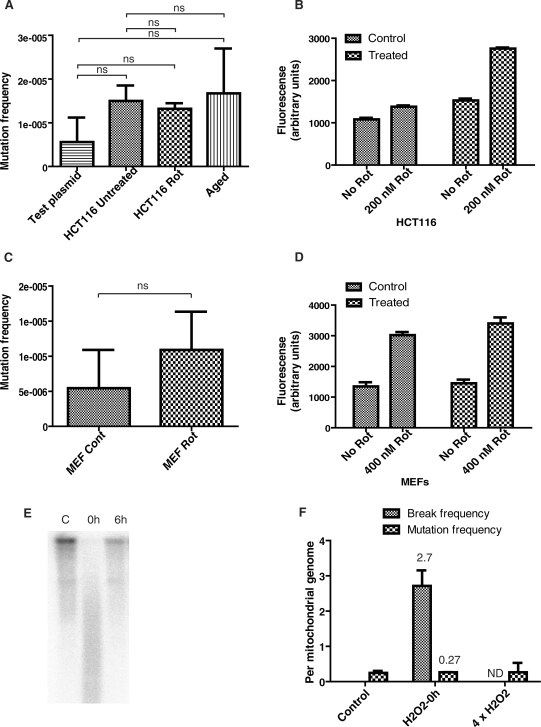
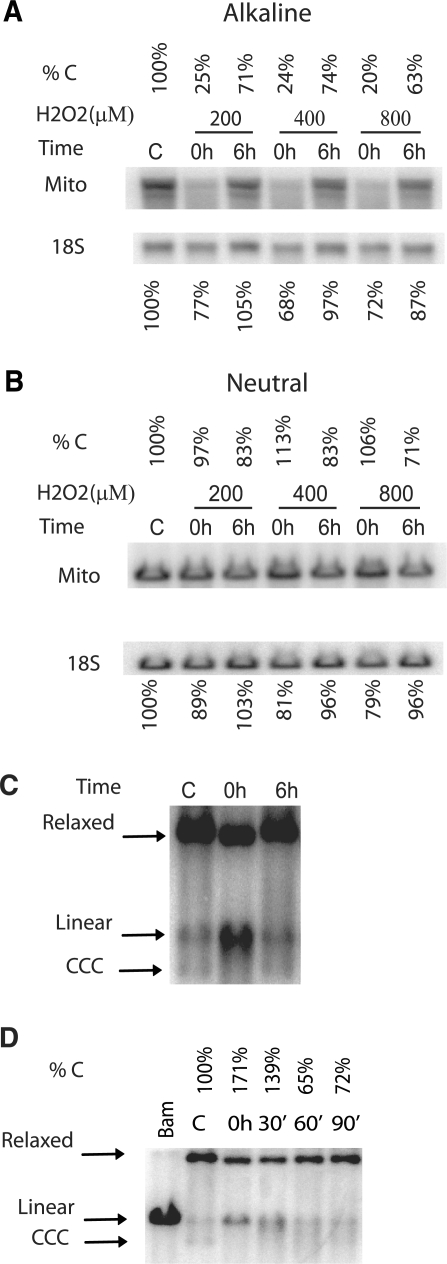
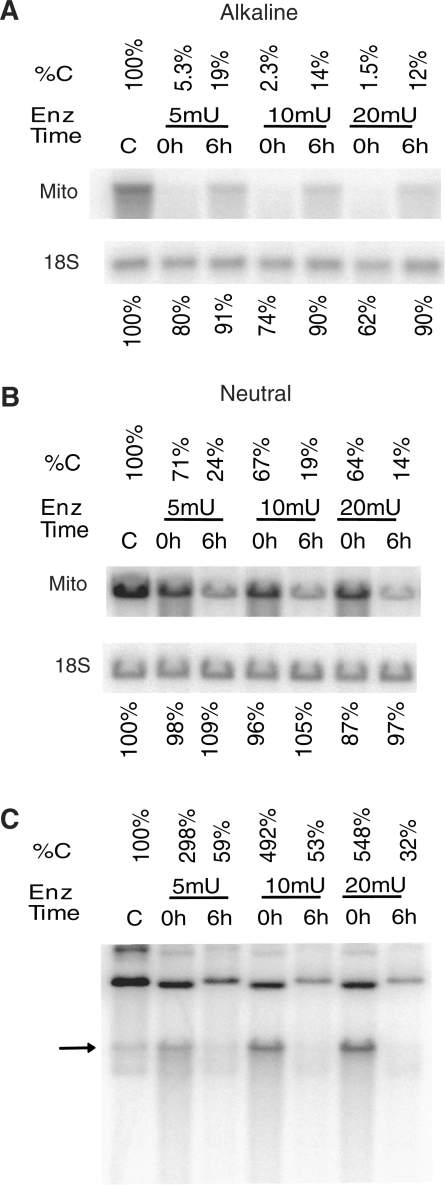
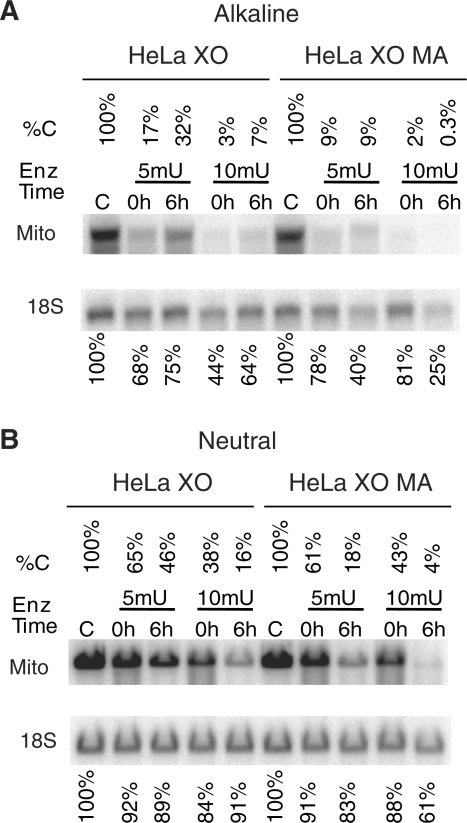
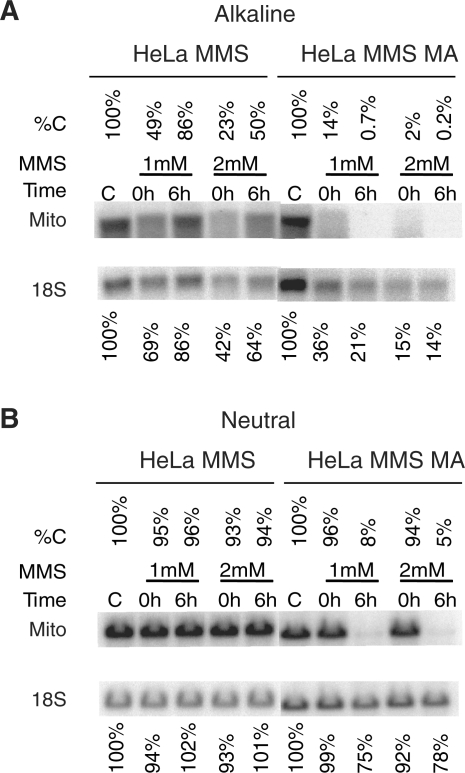
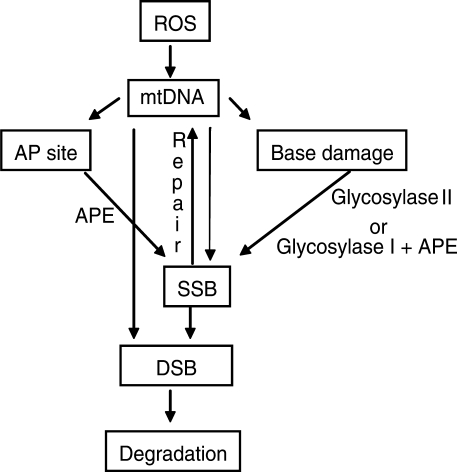
Mitochondrial DNA (mtDNA) is located in close proximity of the respiratory chains, which are the main cellular source of reactive oxygen species (ROS). ROS can induce oxidative base lesions in mtDNA and are believed to be an important cause of the mtDNA mutations, which accumulate with aging and in diseased states. However, recent studies indicate that cumulative levels of base substitutions in mtDNA can be very low even in old individuals. Considering the reduced complement of DNA repair pathways available in mitochondria and higher susceptibility of mtDNA to oxidative damage than nDNA, it is presently unclear how mitochondria manage to maintain the integrity of their genetic information in the face of the permanent exposure to ROS. Here we show that oxidative stress can lead to the degradation of mtDNA and that strand breaks and abasic sites prevail over mutagenic base lesions in ROS-damaged mtDNA. Furthermore, we found that inhibition of base excision repair enhanced mtDNA degradation in response to both oxidative and alkylating damage. These observations suggest a novel mechanism for the protection of mtDNA against oxidative insults whereby a higher incidence of lesions to the sugar-phosphate backbone induces degradation of damaged mtDNA and prevents the accumulation of mutagenic base lesions.
Figures
References
-
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd., Nikoskelainen EK. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. - PubMed
-
- Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. - PubMed
-
- Ohta S. Contribution of somatic mutations in the mitochondrial genome to the development of cancer and tolerance against anticancer drugs. Oncogene. 2006;25:4768–4776. - PubMed
-
- Wallace DC, Lott MT, Shoffner JM, Ballinger S. Mitochondrial DNA mutations in epilepsy and neurological disease. Epilepsia. 1994;35(Suppl. 1):S43–S50. - PubMed
-
- Krishnan KJ, Reeve AK, Turnbull DM. Do mitochondrial DNA mutations have a role in neurodegenerative disease? Biochem. Soc. Trans. 2007;35:1232–1235. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
