

Pyrosequencing of the chaperonin-60 universal target as a tool for determining microbial community composition
- PMID: 19270139
- PMCID: PMC2681704
- DOI: 10.1128/AEM.01640-08
Pyrosequencing of the chaperonin-60 universal target as a tool for determining microbial community composition
Abstract
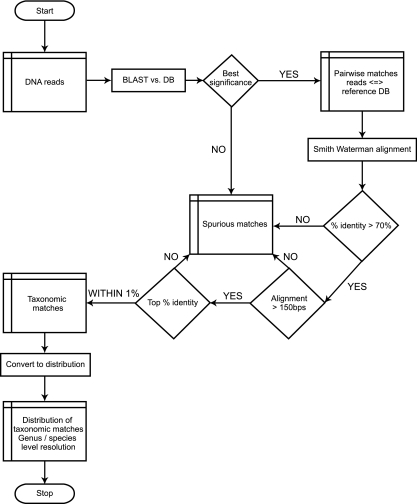
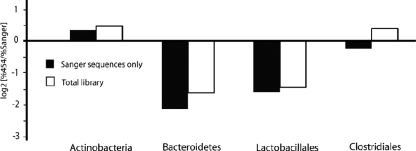
We compared dideoxy sequencing of cloned chaperonin-60 universal target (cpn60 UT) amplicons to pyrosequencing of amplicons derived from vaginal microbial communities. In samples pooled from a number of individuals, the pyrosequencing method produced a data set that included virtually all of the sequences that were found within the clone library and revealed an additional level of taxonomic richness. However, the relative abundances of the sequences were different in the two datasets. These observations were expanded and confirmed by the analysis of paired clone library and pyrosequencing datasets from vaginal swabs taken from four individuals. Both for individuals with a normal vaginal microbiota and for those with bacterial vaginosis, the pyrosequencing method revealed a large number of low-abundance taxa that were missed by the clone library approach. In addition, we showed that the pyrosequencing method generates a reproducible profile of microbial community structure in replicate amplifications from the same community. We also compared the taxonomic composition of a vaginal microbial community determined by pyrosequencing of 16S rRNA amplicons to that obtained using cpn60 universal primers. We found that the profiles generated by the two molecular targets were highly similar, with slight differences in the proportional representation of the taxa detected. However, the number of operational taxonomic units was significantly higher in the cpn60 data set, suggesting that the protein-encoding gene provides improved species resolution over the 16S rRNA target. These observations demonstrate that pyrosequencing of cpn60 UT amplicons provides a robust, reliable method for deep sequencing of microbial communities.
Figures





References
-
- Chou, H. H., and M. H. Holmes. 2001. DNA sequence quality trimming and vector removal. Bioinformatics 17:1093-1104. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
