

Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis
- PMID: 19273622
- PMCID: PMC2699137
- DOI: 10.1084/jem.20081859
Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis
Abstract
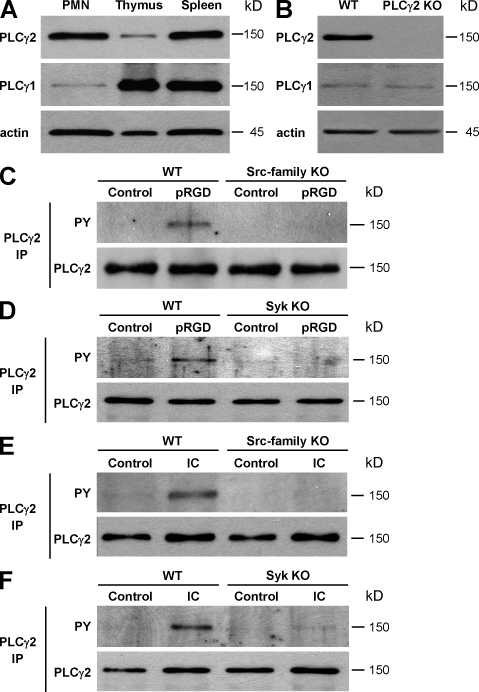
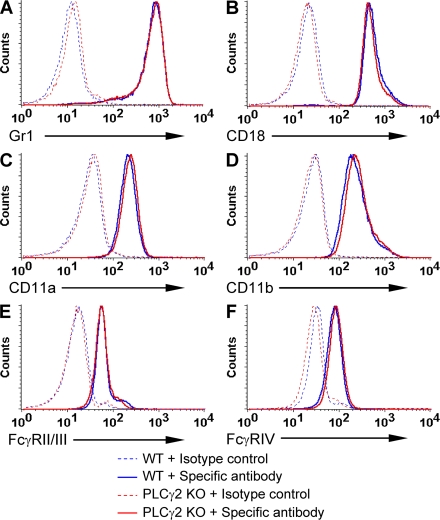
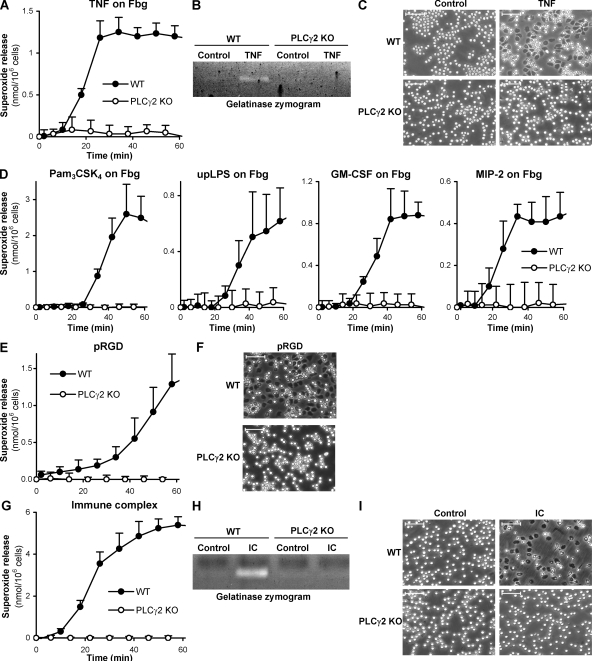
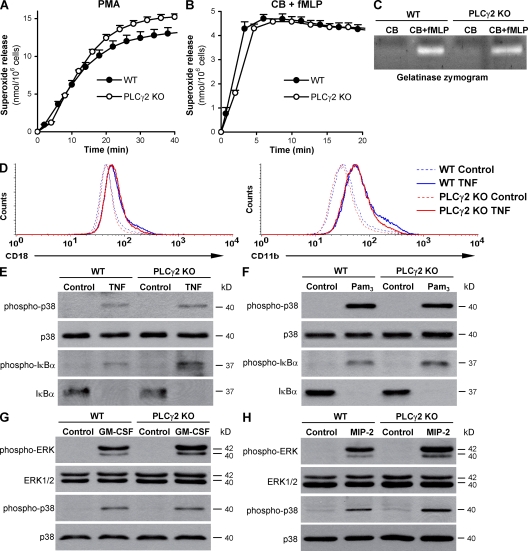
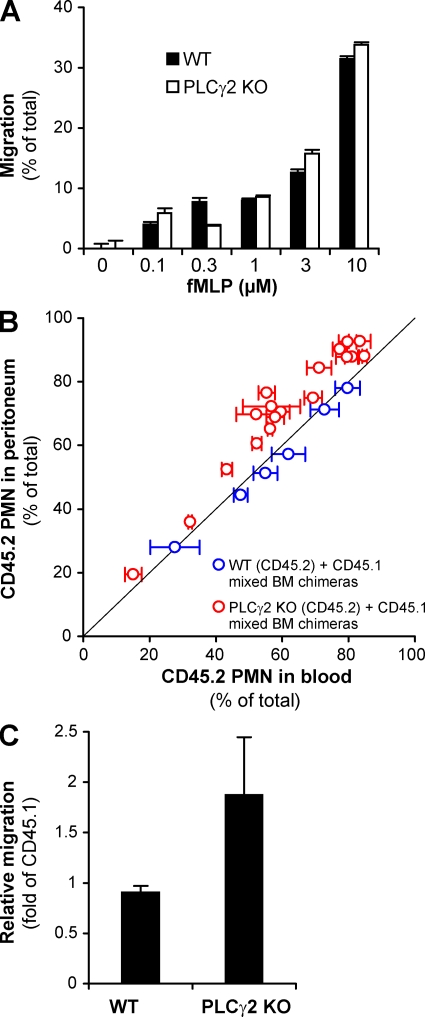
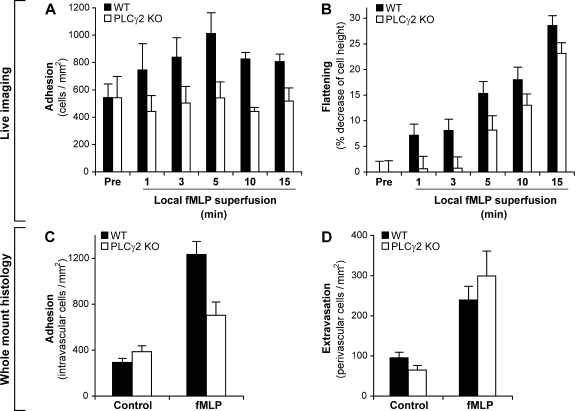
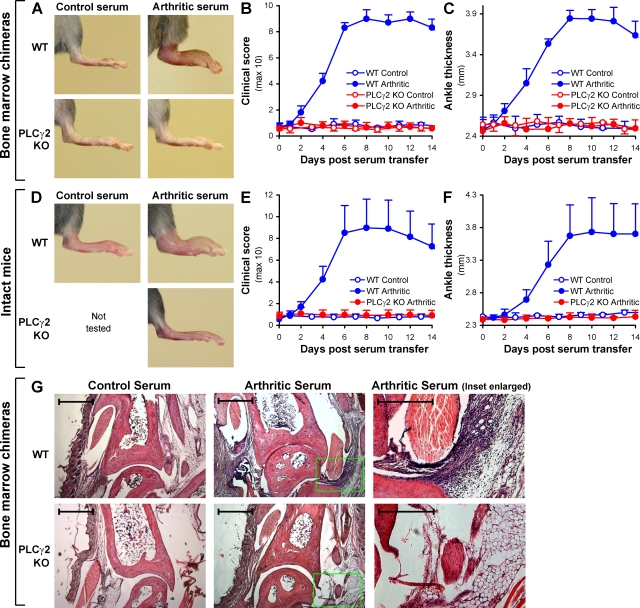
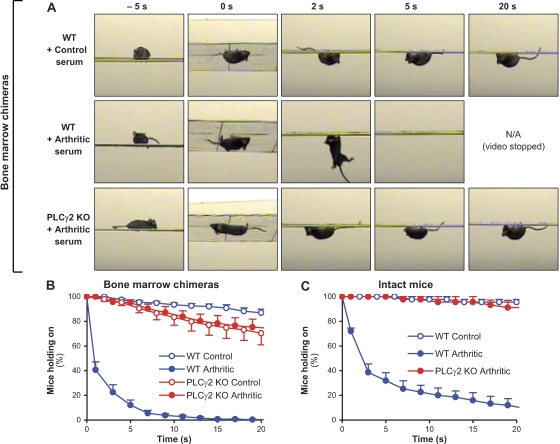
beta(2) integrins and Fcgamma receptors are critically involved in neutrophil activation at the site of inflammation. Both receptor types trigger a receptor-proximal tyrosine phosphorylation cascade through Src family kinases and Syk, but further downstream signaling events are poorly understood. We show that phospholipase C (PLC) gamma2 is phosphorylated downstream of Src family kinases and Syk during integrin or Fc receptor-mediated activation of neutrophils. PLCgamma2(-/-) neutrophils are completely defective in beta(2) integrin or Fcgamma receptor-mediated functional responses such as respiratory burst, degranulation, or cell spreading in vitro and show reduced adhesion/spreading in inflamed capillary venules in vivo. However, PLCgamma2(-/-) neutrophils respond normally to various other agonists, including chemokines, bacterial formyl peptides, Toll-like receptor ligands, or proinflammatory cytokines, and migrate normally both in vitro and in vivo. To confirm the in vivo relevance of these observations, the effect of the PLCgamma2(-/-) mutation was tested in the K/BxN serum transfer arthritis model, which is known to require beta(2) integrins, Fcgamma receptors, and neutrophils. PLCgamma2 deficiency completely protected mice from clinical signs and histological features of arthritis as well as from arthritis-induced loss of articular function. These results identify PLCgamma2 as a critical player of integrin and Fc receptor-mediated neutrophil functions and the neutrophil-mediated effector phase of autoimmune arthritis.
Figures
References
-
- Nathan C. 2006. Neutrophils and immunity: challenges and opportunities.Nat. Rev. Immunol. 6:173–182 - PubMed
-
- Weiss S.J. 1989. Tissue destruction by neutrophils.N. Engl. J. Med. 320:365–376 - PubMed
-
- Eyles J.L., Roberts A.W., Metcalf D., Wicks I.P. 2006. Granulocyte colony-stimulating factor and neutrophils - forgotten mediators of inflammatory disease.Nat. Clin. Pract. Rheumatol. 2:500–510 - PubMed
-
- Nathan C. 2002. Points of control in inflammation.Nature. 420:846–852 - PubMed
-
- Witko-Sarsat V., Rieu P., Descamps-Latscha B., Lesavre P., Halbwachs-Mecarelli L. 2000. Neutrophils: molecules, functions and pathophysiological aspects.Lab. Invest. 80:617–653 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
