

In silico simulation of corticosteroids effect on an NFkB- dependent physicochemical model of systemic inflammation
- PMID: 19274080
- PMCID: PMC2651450
- DOI: 10.1371/journal.pone.0004706
In silico simulation of corticosteroids effect on an NFkB- dependent physicochemical model of systemic inflammation
Abstract
Background: During the onset of an inflammatory response signaling pathways are activated for "translating" extracellular signals into intracellular responses converging to the activation of nuclear factor (NF)-kB, a central transcription factor in driving the inflammatory response. An inadequate control of its transcriptional activity is associated with the culmination of a hyper-inflammatory response making it a desired therapeutic target. Predicated upon the nature of the response, a systems level analysis might provide rational leads for the development of strategies that promote the resolution of the response.
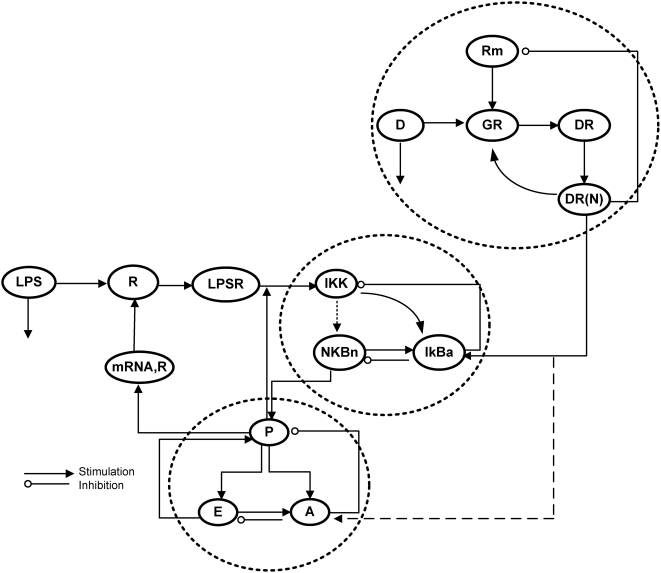
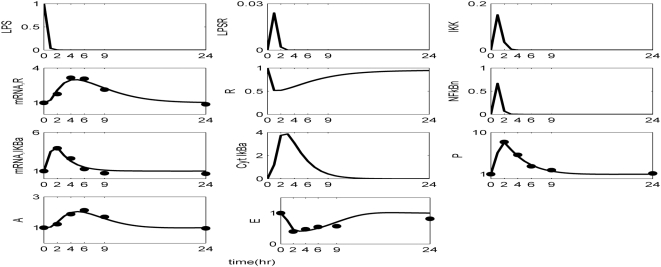
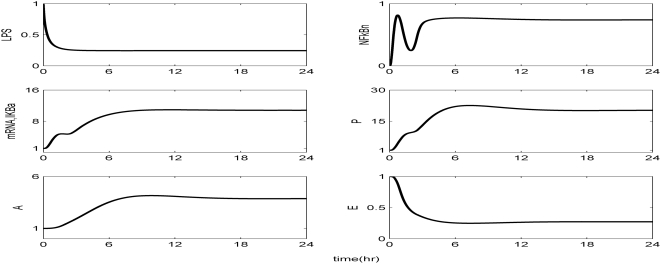
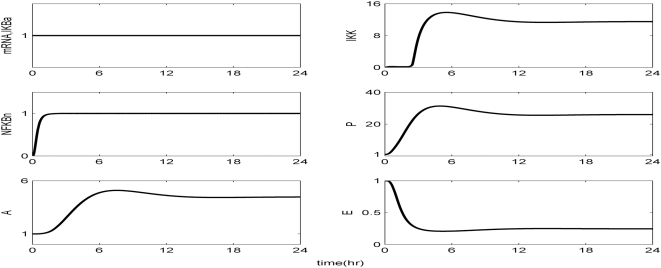
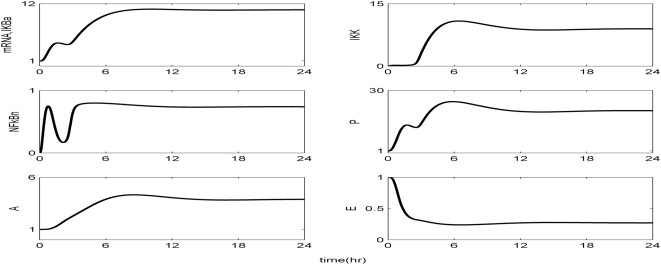
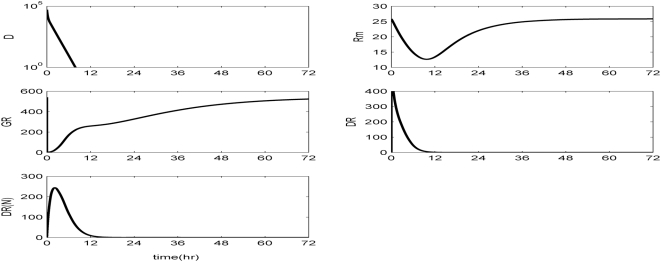
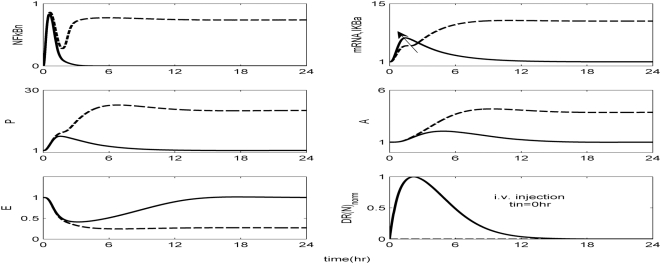
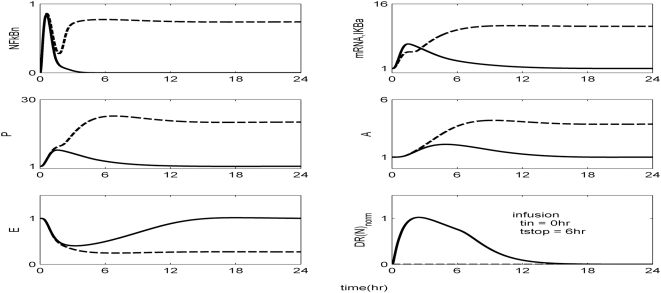
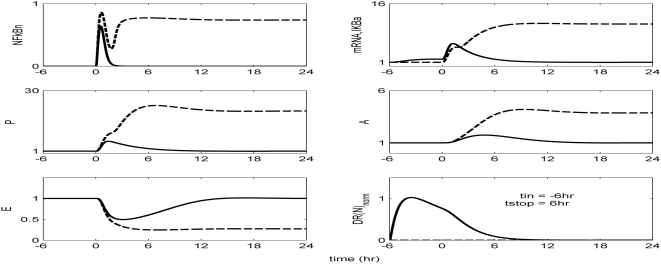
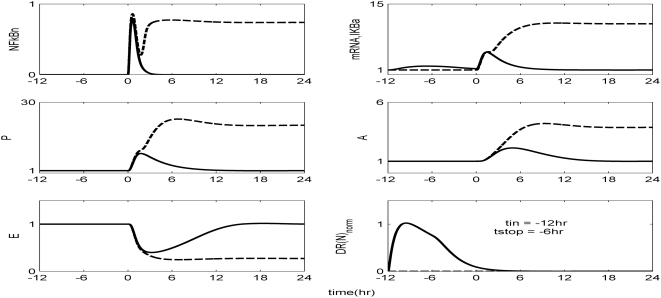
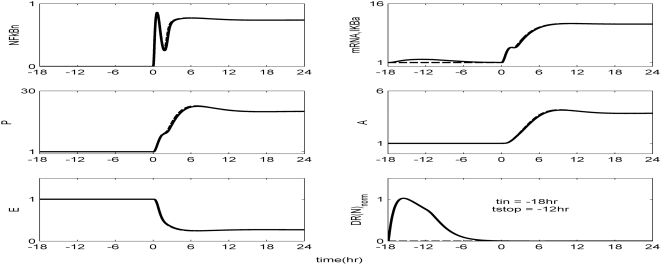
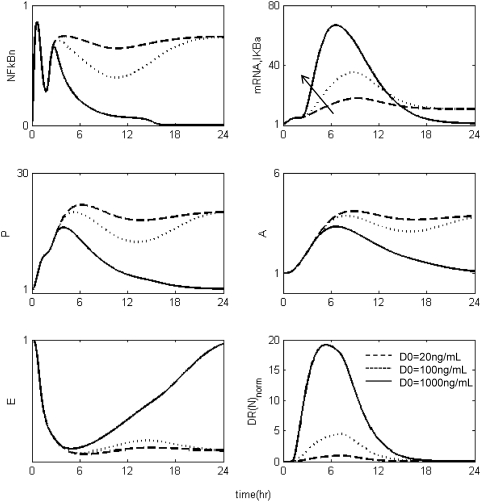
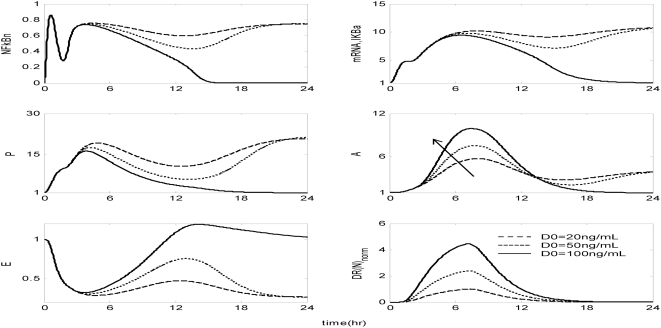
Methodology and findings: A physicochemical host response model is proposed to integrate biological information in the form of kinetic rules and signaling cascades with pharmacokinetic models of drug action for the modulation of the response. The unifying hypothesis is that the response is triggered by the activation of the NFkB signaling module and corticosteroids serve as a template for assessing anti-inflammatory strategies. The proposed in silico model is evaluated through its ability to predict and modulate uncontrolled responses. The pre-exposure of the system to hypercortisolemia, i.e. 6 hr before or simultaneously with the infectious challenge "reprograms" the dynamics of the host towards a balanced inflammatory response. However, if such an intervention occurs long before the inflammatory insult a symptomatic effect is observed instead of a protective relief while a steroid infusion after inducing inflammation requires much higher drug doses.
Conclusions and significance: We propose a reversed engineered inflammation model that seeks to describe how the system responds to a multitude of external signals. Timing of intervention and dosage regimes appears to be key determinants for the protective or symptomatic effect of exogenous corticosteroids. Such results lie in qualitative agreement with in vivo human studies exposed both to LPS and corticosteroids under various time intervals thus improving our understanding of how interacting modules generate a behavior.
Conflict of interest statement
Figures
Similar articles
-
Corticosteroids alleviate lipopolysaccharide-induced inflammation and lung injury via inhibiting NLRP3-inflammasome activation.J Cell Mol Med. 2020 Nov;24(21):12716-12725. doi: 10.1111/jcmm.15849. Epub 2020 Sep 25. J Cell Mol Med. 2020. PMID: 32977368 Free PMC article.
-
Agent-based modeling of endotoxin-induced acute inflammatory response in human blood leukocytes.PLoS One. 2010 Feb 18;5(2):e9249. doi: 10.1371/journal.pone.0009249. PLoS One. 2010. PMID: 20174629 Free PMC article.
-
Computational modeling with forward and reverse engineering links signaling network and genomic regulatory responses: NF-kappaB signaling-induced gene expression responses in inflammation.BMC Bioinformatics. 2010 Jun 8;11:308. doi: 10.1186/1471-2105-11-308. BMC Bioinformatics. 2010. PMID: 20529327 Free PMC article.
-
Nuclear factor kappa B: a pivotal role in the systemic inflammatory response syndrome and new target for therapy.Intensive Care Med. 1998 Nov;24(11):1131-8. doi: 10.1007/s001340050735. Intensive Care Med. 1998. PMID: 9876974 Free PMC article. Review.
-
Corticosteroids: the drugs to beat.Eur J Pharmacol. 2006 Mar 8;533(1-3):2-14. doi: 10.1016/j.ejphar.2005.12.052. Epub 2006 Jan 24. Eur J Pharmacol. 2006. PMID: 16436275 Review.
Cited by
-
Translational applications of evaluating physiologic variability in human endotoxemia.J Clin Monit Comput. 2013 Aug;27(4):405-15. doi: 10.1007/s10877-012-9418-1. Epub 2012 Dec 1. J Clin Monit Comput. 2013. PMID: 23203205 Free PMC article. Review.
-
In Silico Augmentation of the Drug Development Pipeline: Examples from the study of Acute Inflammation.Drug Dev Res. 2011 Mar 1;72(2):187-200. doi: 10.1002/ddr.20415. Drug Dev Res. 2011. PMID: 21552346 Free PMC article.
-
Lipopolysaccharide-induced inhibition of transcription of tlr4 in vitro is reversed by dexamethasone and correlates with presence of conserved NFκB binding sites.Biochem Biophys Res Commun. 2013 Mar 8;432(2):256-61. doi: 10.1016/j.bbrc.2013.02.002. Epub 2013 Feb 10. Biochem Biophys Res Commun. 2013. PMID: 23402753 Free PMC article.
-
Pharmacokinetic/pharmacodynamic modeling in inflammation.Crit Rev Biomed Eng. 2012;40(4):295-312. doi: 10.1615/critrevbiomedeng.v40.i4.50. Crit Rev Biomed Eng. 2012. PMID: 23140121 Free PMC article. Review.
-
A Systems Engineering Perspective on Homeostasis and Disease.Front Bioeng Biotechnol. 2013 Sep 9;1:6. doi: 10.3389/fbioe.2013.00006. eCollection 2013. Front Bioeng Biotechnol. 2013. PMID: 25022216 Free PMC article. Review.
References
-
- Nystrom PO. The systemic inflammatory response syndrome: definitions and aetiology. J Antimicrob Chemother. 1998;41(Suppl A):1–7. - PubMed
-
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. - PubMed
-
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. - PubMed
-
- Tetta C, Fonsato V, Ronco C, Camussi G. Recent insights into the pathogenesis of severe sepsis. Crit Care Resusc. 2005;7:32–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
