

Epitope-tagged receptor knock-in mice reveal that differential desensitization of alpha2-adrenergic responses is because of ligand-selective internalization
- PMID: 19276088
- PMCID: PMC2676055
- DOI: 10.1074/jbc.M807535200
Epitope-tagged receptor knock-in mice reveal that differential desensitization of alpha2-adrenergic responses is because of ligand-selective internalization
Abstract
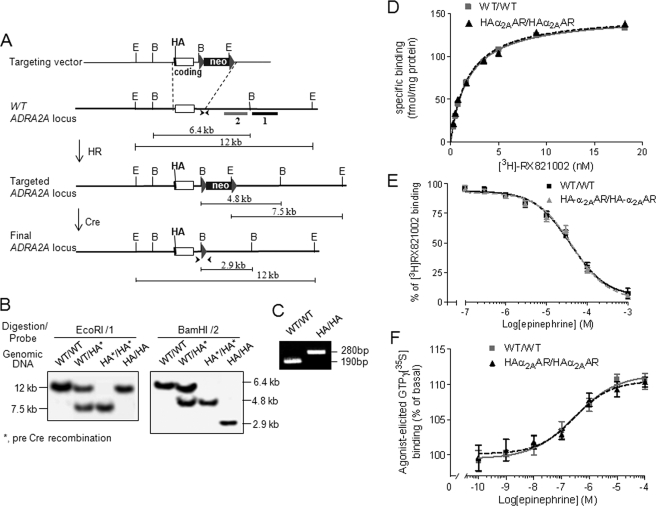
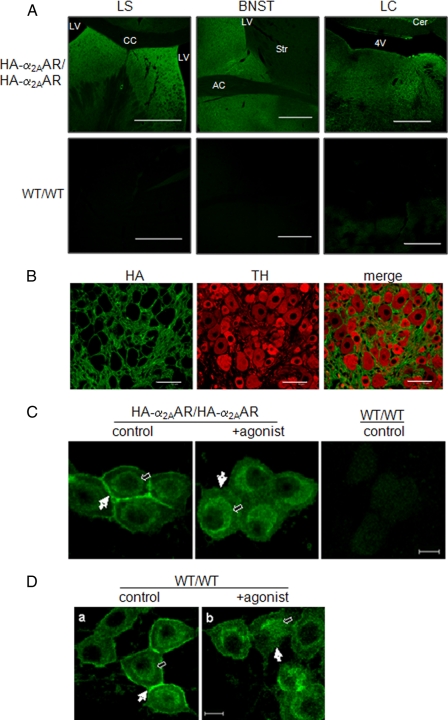
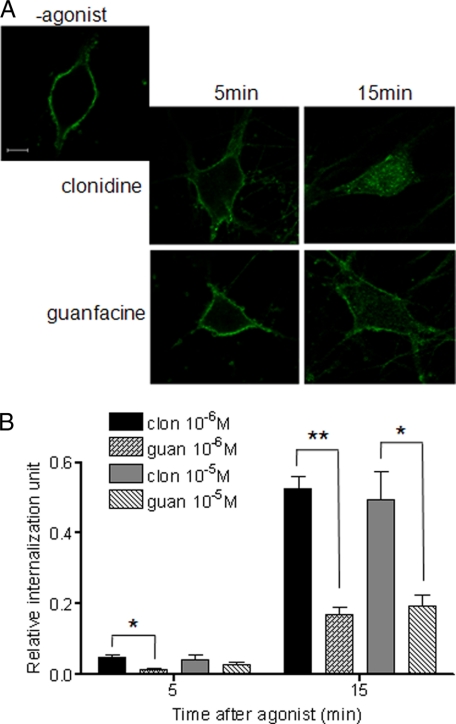
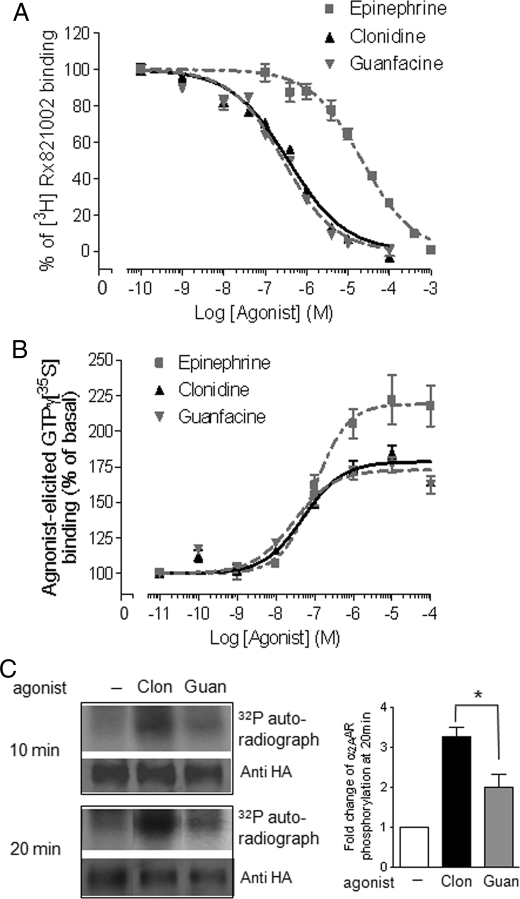
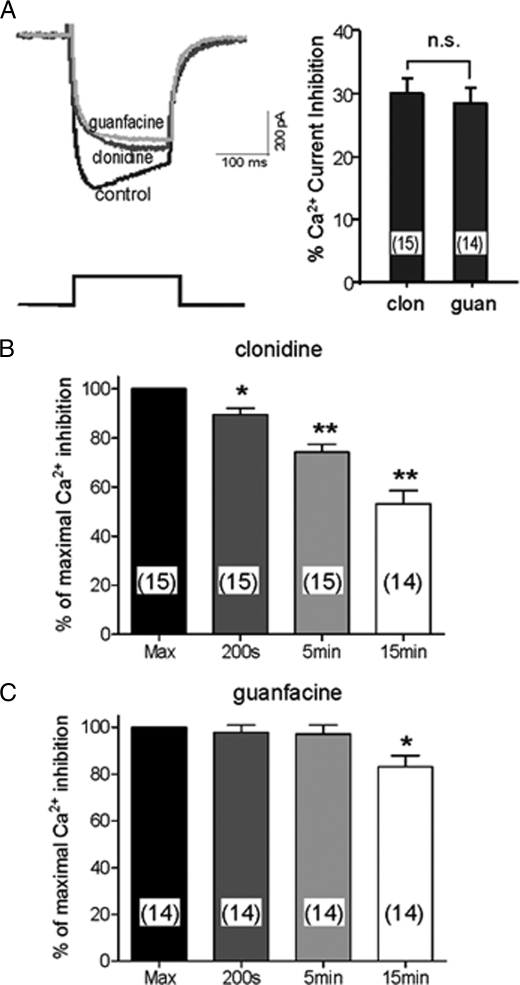
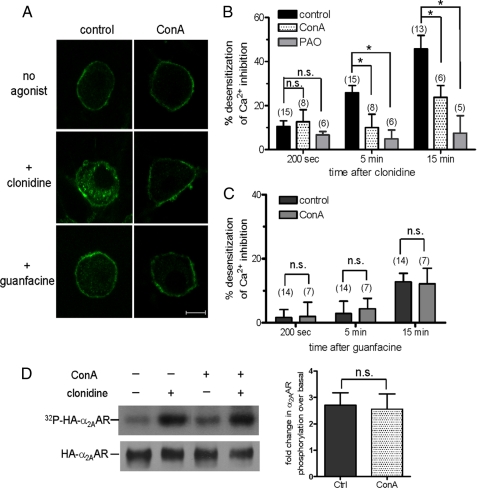
Although ligand-selective regulation of G protein-coupled receptor-mediated signaling and trafficking are well documented, little is known about whether ligand-selective effects occur on endogenous receptors or whether such effects modify the signaling response in physiologically relevant cells. Using a gene targeting approach, we generated a knock-in mouse line, in which N-terminal hemagglutinin epitope-tagged alpha(2A)-adrenergic receptor (AR) expression was driven by the endogenous mouse alpha(2A)AR gene locus. Exploiting this mouse line, we evaluated alpha(2A)AR trafficking and alpha(2A)AR-mediated inhibition of Ca(2+) currents in native sympathetic neurons in response to clonidine and guanfacine, two drugs used for treatment of hypertension, attention deficit and hyperactivity disorder, and enhancement of analgesia through actions on the alpha(2A)AR subtype. We discovered a more rapid desensitization of Ca(2+) current suppression by clonidine than guanfacine, which paralleled a more marked receptor phosphorylation and endocytosis of alpha(2A)AR evoked by clonidine than by guanfacine. Clonidine-induced alpha(2A)AR desensitization, but not receptor phosphorylation, was attenuated by blockade of endocytosis with concanavalin A, indicating a critical role for internalization of alpha(2A)AR in desensitization to this ligand. Our data on endogenous receptor-mediated signaling and trafficking in native cells reveal not only differential regulation of G protein-coupled receptor endocytosis by different ligands, but also a differential contribution of receptor endocytosis to signaling desensitization. Taken together, our data suggest that these HA-alpha(2A)AR knock-in mice will serve as an important model in developing ligands to favor endocytosis or nonendocytosis of receptors, depending on the target cell and pathophysiology being addressed.
Figures
Similar articles
-
alpha2A-adrenergic receptors heterosynaptically regulate glutamatergic transmission in the bed nucleus of the stria terminalis.Neuroscience. 2009 Sep 29;163(1):339-51. doi: 10.1016/j.neuroscience.2009.06.022. Epub 2009 Jun 12. Neuroscience. 2009. PMID: 19527774 Free PMC article.
-
alpha(2C)-Adrenergic receptors mediate spinal analgesia and adrenergic-opioid synergy.J Pharmacol Exp Ther. 2002 Jan;300(1):282-90. doi: 10.1124/jpet.300.1.282. J Pharmacol Exp Ther. 2002. PMID: 11752127
-
The anticonvulsant and proconvulsant effects of alpha2-adrenoreceptor agonists are mediated by distinct populations of alpha2A-adrenoreceptors.Neuroscience. 2004;126(3):795-803. doi: 10.1016/j.neuroscience.2004.04.030. Neuroscience. 2004. PMID: 15183527
-
The contribution of alpha 2-noradrenergic mechanisms of prefrontal cortical cognitive function. Potential significance for attention-deficit hyperactivity disorder.Arch Gen Psychiatry. 1996 May;53(5):448-55. doi: 10.1001/archpsyc.1996.01830050084013. Arch Gen Psychiatry. 1996. PMID: 8624188 Review.
-
[Adrenergic receptor and knockout mouse: 2). Alpha adrenergic receptor knockout mouse].Masui. 2001 Jan;50(1):12-9. Masui. 2001. PMID: 11211741 Review. Japanese.
Cited by
-
An N-terminal fusion allele to study melanin concentrating hormone receptor 1.Genesis. 2021 Aug;59(7-8):e23438. doi: 10.1002/dvg.23438. Epub 2021 Jun 14. Genesis. 2021. PMID: 34124835 Free PMC article.
-
α(2A)-adrenergic receptors filter parabrachial inputs to the bed nucleus of the stria terminalis.J Neurosci. 2014 Jul 9;34(28):9319-31. doi: 10.1523/JNEUROSCI.0822-14.2014. J Neurosci. 2014. PMID: 25009265 Free PMC article.
-
Cellular Heterogeneity of the Luteinizing Hormone Receptor and Its Significance for Cyclic GMP Signaling in Mouse Preovulatory Follicles.Endocrinology. 2020 Jul 1;161(7):bqaa074. doi: 10.1210/endocr/bqaa074. Endocrinology. 2020. PMID: 32384146 Free PMC article.
-
The antidepressant desipramine is an arrestin-biased ligand at the α(2A)-adrenergic receptor driving receptor down-regulation in vitro and in vivo.J Biol Chem. 2011 Oct 14;286(41):36063-36075. doi: 10.1074/jbc.M111.261578. Epub 2011 Aug 22. J Biol Chem. 2011. PMID: 21859713 Free PMC article.
-
Knock-In Mouse Models to Investigate the Functions of Opioid Receptors in vivo.Front Cell Neurosci. 2022 Jan 31;16:807549. doi: 10.3389/fncel.2022.807549. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35173584 Free PMC article. Review.
References
-
- Pierce, K. L., Premont, R. T., and Lefkowitz, R. J. (2002) Nat. Rev. Mol. Cell Biol. 3 639-650 - PubMed
-
- Kohout, T. A., and Lefkowitz, R. J. (2003) Mol. Pharmacol. 63 9-18 - PubMed
-
- Gainetdinov, R. R., Premont, R. T., Bohn, L. M., Lefkowitz, R. J., and Caron, M. G. (2004) Annu. Rev. Neurosci. 27 107-144 - PubMed
-
- Pierce, K. L., and Lefkowitz, R. J. (2001) Nat. Rev. Neurosci. 2 727-733 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
