

Correction for phylogeny, small number of observations and data redundancy improves the identification of coevolving amino acid pairs using mutual information
- PMID: 19276150
- PMCID: PMC2672635
- DOI: 10.1093/bioinformatics/btp135
Correction for phylogeny, small number of observations and data redundancy improves the identification of coevolving amino acid pairs using mutual information
Abstract
Motivation: Mutual information (MI) theory is often applied to predict positional correlations in a multiple sequence alignment (MSA) to make possible the analysis of those positions structurally or functionally important in a given fold or protein family. Accurate identification of coevolving positions in protein sequences is difficult due to the high background signal imposed by phylogeny and noise. Several methods have been proposed using MI to identify coevolving amino acids in protein families.
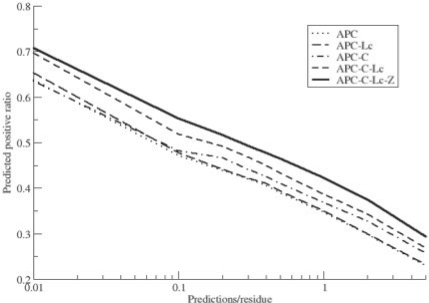
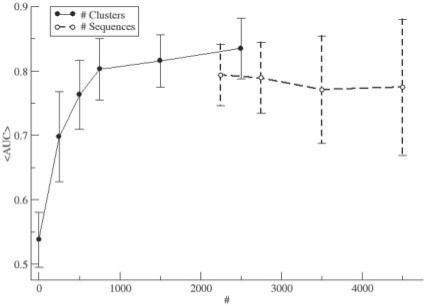
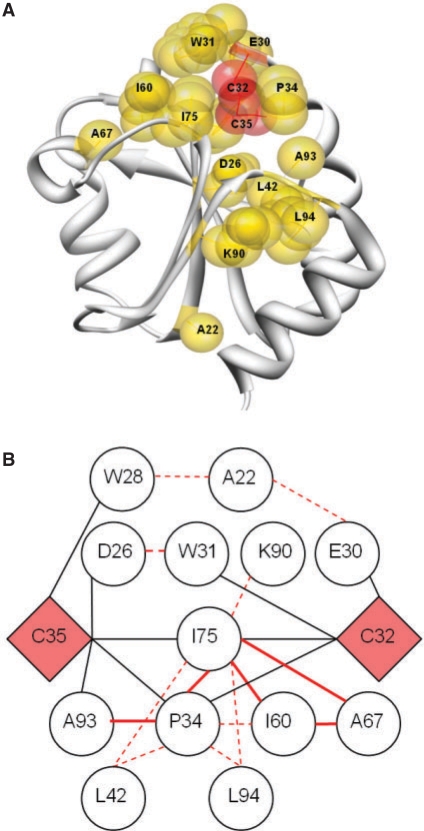
Results: After evaluating two current methods, we demonstrate how the use of sequence-weighting techniques to reduce sequence redundancy and low-count corrections to account for small number of observations in limited size sequence families, can significantly improve the predictability of MI. The evaluation is made on large sets of both in silico-generated alignments as well as on biological sequence data. The methods included in the analysis are the APC (average product correction) and RCW (row-column weighting) methods. The best performing method was APC including sequence-weighting and low-count corrections. The use of sequence-permutations to calculate a MI rescaling is shown to significantly improve the prediction accuracy and allows for direct comparison of information values across protein families. Finally, we demonstrate how a lower bound of 400 sequences <62% identical is needed in an MSA in order to achieve meaningful predictive performances. With our contribution, we achieve a noteworthy improvement on the current procedures to determine coevolution and residue contacts, and we believe that this will have potential impacts on the understanding of protein structure, function and folding.
Figures
Similar articles
-
Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction.Bioinformatics. 2008 Feb 1;24(3):333-40. doi: 10.1093/bioinformatics/btm604. Epub 2007 Dec 5. Bioinformatics. 2008. PMID: 18057019
-
Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions.Biochemistry. 2005 May 17;44(19):7156-65. doi: 10.1021/bi050293e. Biochemistry. 2005. PMID: 15882054
-
Using information theory to search for co-evolving residues in proteins.Bioinformatics. 2005 Nov 15;21(22):4116-24. doi: 10.1093/bioinformatics/bti671. Epub 2005 Sep 13. Bioinformatics. 2005. PMID: 16159918
-
A practical guide for the computational selection of residues to be experimentally characterized in protein families.Brief Bioinform. 2012 May;13(3):329-36. doi: 10.1093/bib/bbr052. Epub 2011 Sep 19. Brief Bioinform. 2012. PMID: 21930656 Review.
-
Upcoming challenges for multiple sequence alignment methods in the high-throughput era.Bioinformatics. 2009 Oct 1;25(19):2455-65. doi: 10.1093/bioinformatics/btp452. Epub 2009 Jul 30. Bioinformatics. 2009. PMID: 19648142 Free PMC article. Review.
Cited by
-
Structural and functional roles of coevolved sites in proteins.PLoS One. 2010 Jan 6;5(1):e8591. doi: 10.1371/journal.pone.0008591. PLoS One. 2010. PMID: 20066038 Free PMC article.
-
MitImpact 3: modeling the residue interaction network of the Respiratory Chain subunits.Nucleic Acids Res. 2021 Jan 8;49(D1):D1282-D1288. doi: 10.1093/nar/gkaa1032. Nucleic Acids Res. 2021. PMID: 33300029 Free PMC article.
-
I-COMS: Interprotein-COrrelated Mutations Server.Nucleic Acids Res. 2015 Jul 1;43(W1):W320-5. doi: 10.1093/nar/gkv572. Epub 2015 Jun 1. Nucleic Acids Res. 2015. PMID: 26032772 Free PMC article.
-
Chasing coevolutionary signals in intrinsically disordered proteins complexes.Sci Rep. 2020 Oct 21;10(1):17962. doi: 10.1038/s41598-020-74791-6. Sci Rep. 2020. PMID: 33087759 Free PMC article.
-
RNAconTest: comparing tools for noncoding RNA multiple sequence alignment based on structural consistency.RNA. 2020 May;26(5):531-540. doi: 10.1261/rna.073015.119. Epub 2020 Jan 31. RNA. 2020. PMID: 32005745 Free PMC article.
References
-
- Byung-Chul L, et al. Analysis of the residue-residue coevolution network and the functionally important residues in proteins. Protein Struct. Funct. Bioinform. 2008;72:863–872. - PubMed
-
- Chiu DKY, Kolodziejczak T. Inferring consensus structure from nucleic acid sequences. Comput. Appl. Biosci. 1991;7:347–352. - PubMed
-
- Cover TM, Thomas JA. Elements of information theory. Wiley; 1991.
-
- DePristo MA, et al. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 2005;6:678–687. - PubMed
