

Joint estimation of copy number variation and reference intensities on multiple DNA arrays using GADA
- PMID: 19276152
- PMCID: PMC2732310
- DOI: 10.1093/bioinformatics/btp119
Joint estimation of copy number variation and reference intensities on multiple DNA arrays using GADA
Abstract
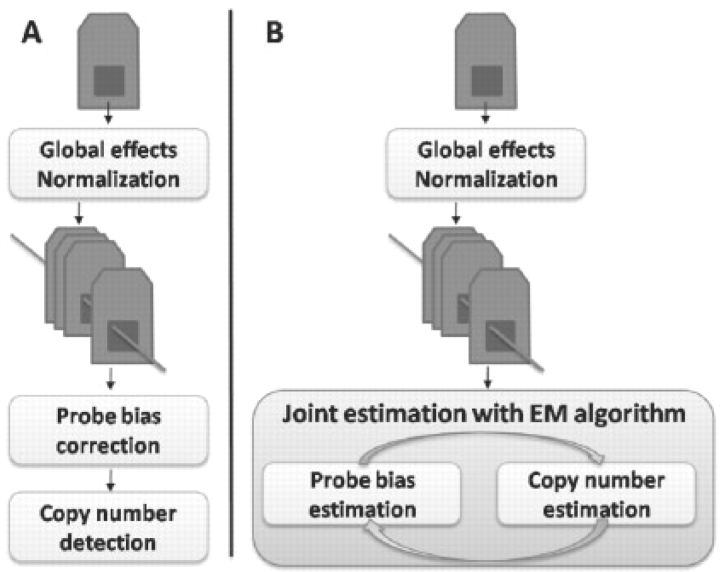
Motivation: The complexity of a large number of recently discovered copy number polymorphisms is much higher than initially thought, thus making it more difficult to detect them in the presence of significant measurement noise. In this scenario, separate normalization and segmentation is prone to lead to many false detections of changes in copy number. New approaches capable of jointly modeling the copy number and the non-copy number (noise) hybridization effects across multiple samples will potentially lead to more accurate results.
Methods: In this article, the genome alteration detection analysis (GADA) approach introduced in our previous work is extended to a multiple sample model. The copy number component is independent for each sample and uses a sparse Bayesian prior, while the reference hybridization level is not necessarily sparse but identical on all samples. The expectation maximization (EM) algorithm used to fit the model iteratively determines whether the observed hybridization levels are more likely due to a copy number variation or to a shared hybridization bias.
Results: The new proposed approach is compared with the currently used strategy of separate normalization followed by independent segmentation of each array. Real microarray data obtained from HapMap samples are randomly partitioned to create different reference sets. Using the new approach, copy number and reference intensity estimates are significantly less variable if the reference set changes; and a higher consistency on copy numbers detected within HapMap family trios is obtained. Finally, the running time to fit the model grows linearly in the number samples and probes.
Availability: http://biron.usc.edu/~piquereg/GADA.
Figures








References
-
- Affymetrix Genome-wide human snp array 6.0 sample data set. 2007 Available at http://www.affymetrix.com/support/technical/sample_data/genomewide_snp6\...(last accessed date February 17, 2009)
-
- Affymetrix Genotyping Console 3.0.1 User Manual. 2008 Available at ftp://www.affymetrix.com/(last accessed date February 17, 2009)
-
- Bengtsson H, et al. Estimation and assessment of raw copy numbers at the single locus level. Bioinformatics. 2008;24:759–767. - PubMed
-
- Feuk L, et al. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. - PubMed
