

CDC25B mediates rapamycin-induced oncogenic responses in cancer cells
- PMID: 19276368
- PMCID: PMC2697620
- DOI: 10.1158/0008-5472.CAN-08-3222
CDC25B mediates rapamycin-induced oncogenic responses in cancer cells
Abstract
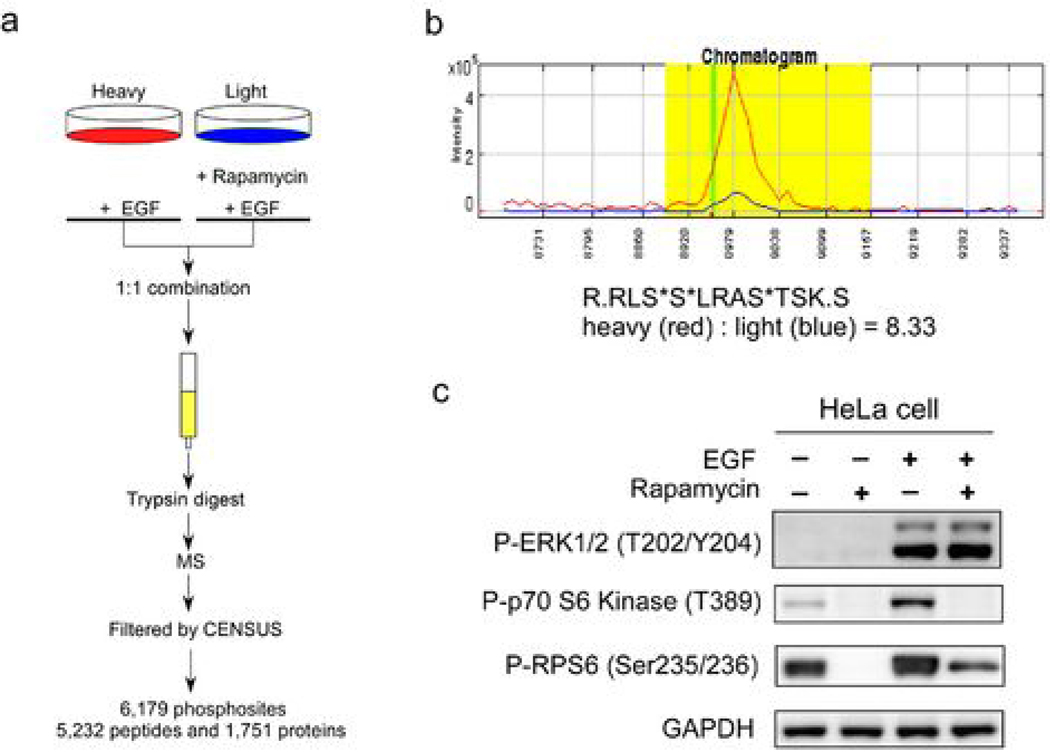
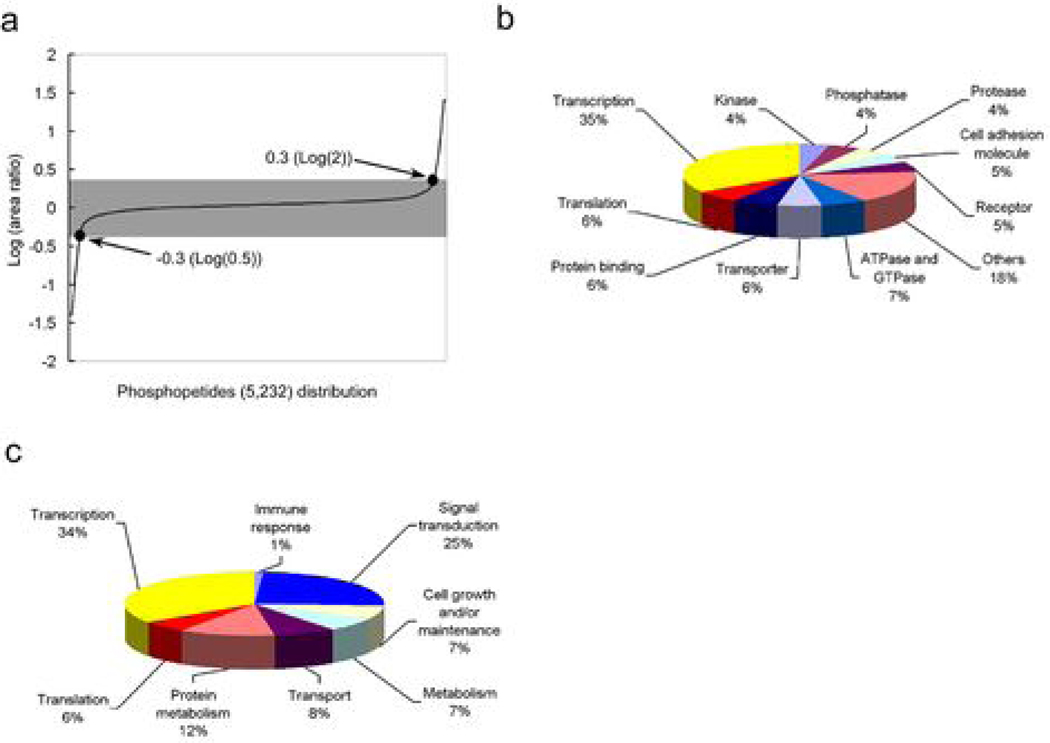
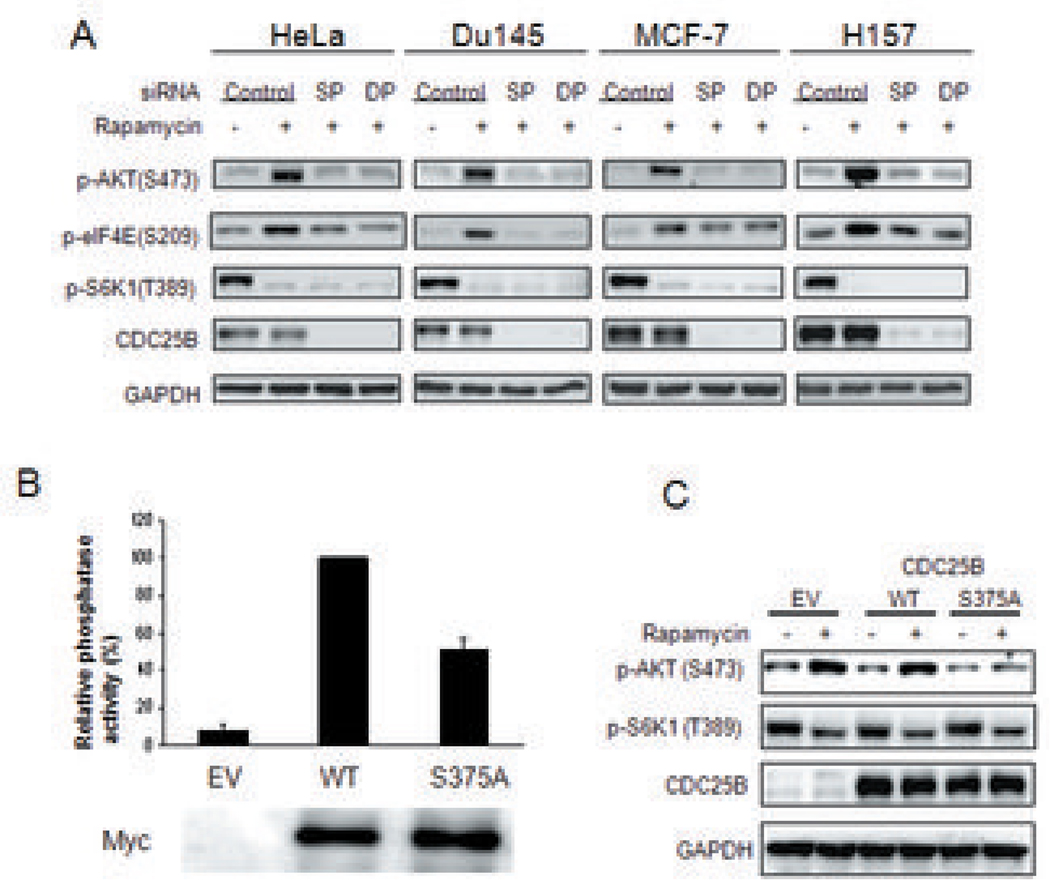
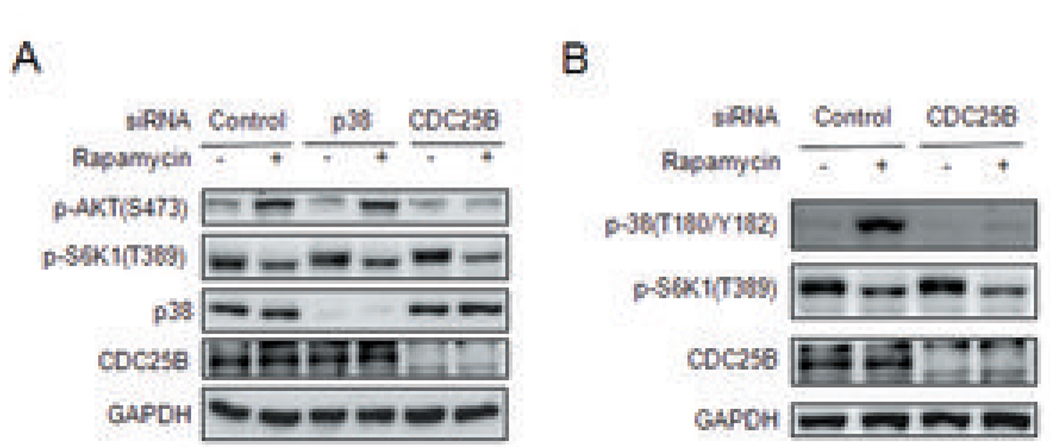
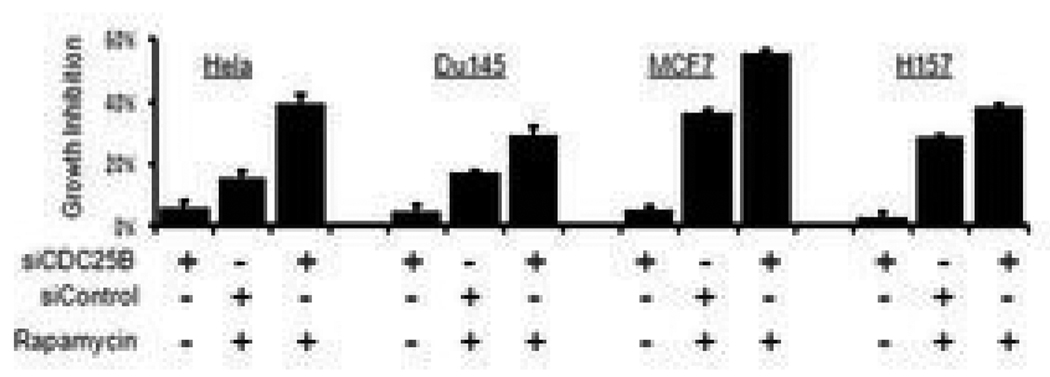
Because the mammalian target of rapamycin (mTOR) pathway is commonly deregulated in human cancer, mTOR inhibitors, rapamycin and its derivatives, are being actively tested in cancer clinical trials. Clinical updates indicate that the anticancer effect of these drugs is limited, perhaps due to rapamycin-dependent induction of oncogenic cascades by an as yet unclear mechanism. As such, we investigated rapamycin-dependent phosphoproteomics and discovered that 250 phosphosites in 161 cellular proteins were sensitive to rapamycin. Among these, rapamycin regulated four kinases and four phosphatases. A siRNA-dependent screen of these proteins showed that AKT induction by rapamycin was attenuated by depleting cellular CDC25B phosphatase. Rapamycin induces the phosphorylation of CDC25B at Serine375, and mutating this site to Alanine substantially reduced CDC25B phosphatase activity. Additionally, expression of CDC25B (S375A) inhibited the AKT activation by rapamycin, indicating that phosphorylation of CDC25B is critical for CDC25B activity and its ability to transduce rapamycin-induced oncogenic AKT activity. Importantly, we also found that CDC25B depletion in various cancer cell lines enhanced the anticancer effect of rapamycin. Together, using rapamycin phosphoproteomics, we not only advance the global mechanistic understanding of the action of rapamycin but also show that CDC25B may serve as a drug target for improving mTOR-targeted cancer therapies.
Figures
References
-
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. - PubMed
-
- Sawyers CL. Will mTOR inhibitors make it as cancer drugs? Cancer Cell. 2003;4:343–348. - PubMed
-
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. - PubMed
-
- Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446. - PubMed
-
- Rampaul RS, Pinder SE, Gullick WJ, Robertson JF, Ellis IO. HER-2 in breast cancer--methods of detection, clinical significance and future prospects for treatment. Crit Rev Oncol Hematol. 2002;43:231–244. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
