

Rhomboid protease dynamics and lipid interactions
- PMID: 19278654
- PMCID: PMC2679947
- DOI: 10.1016/j.str.2008.12.017
Rhomboid protease dynamics and lipid interactions
Abstract
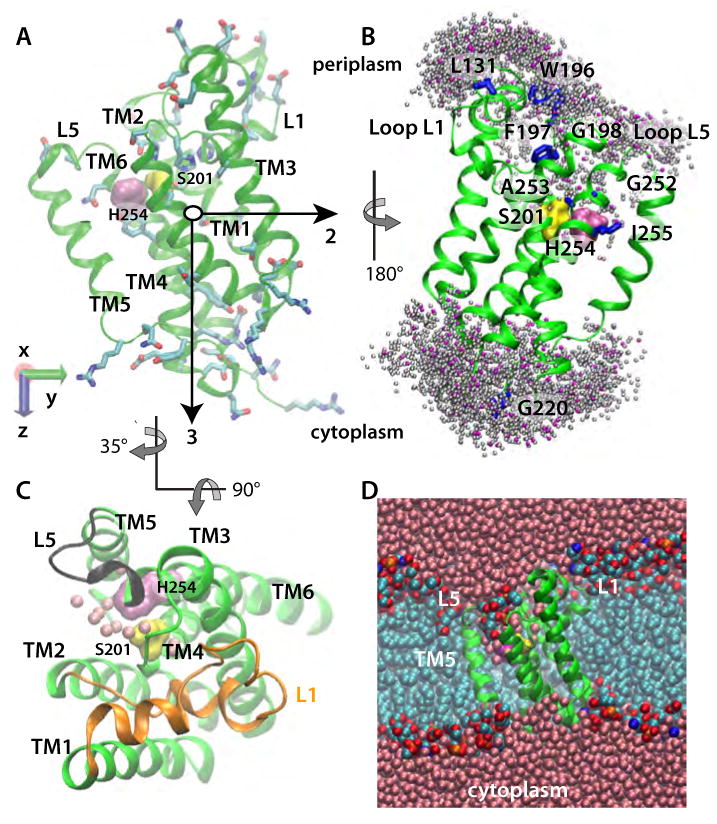
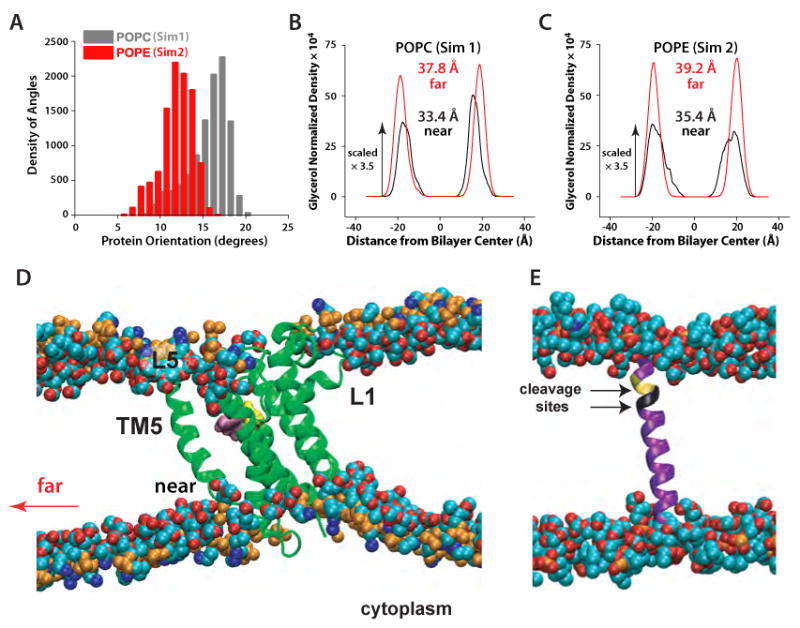
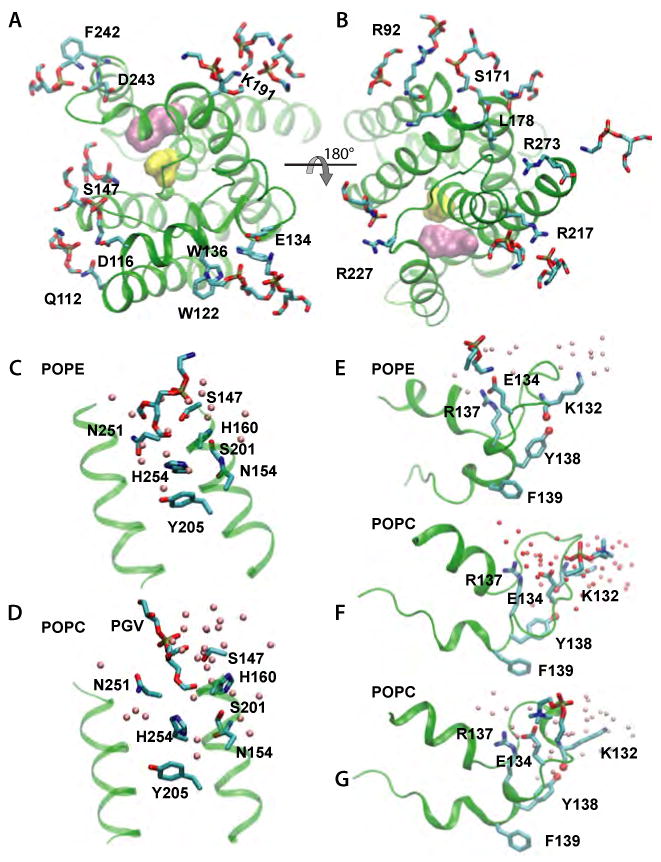
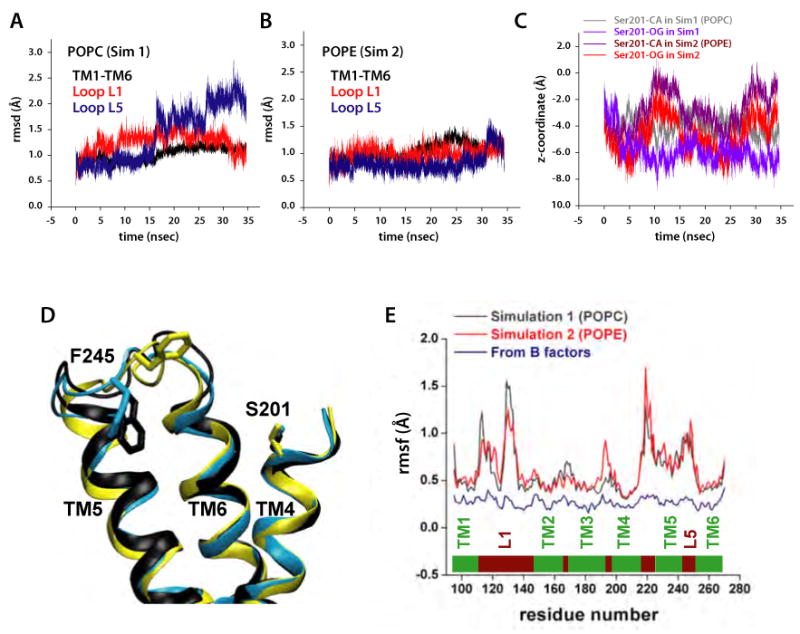
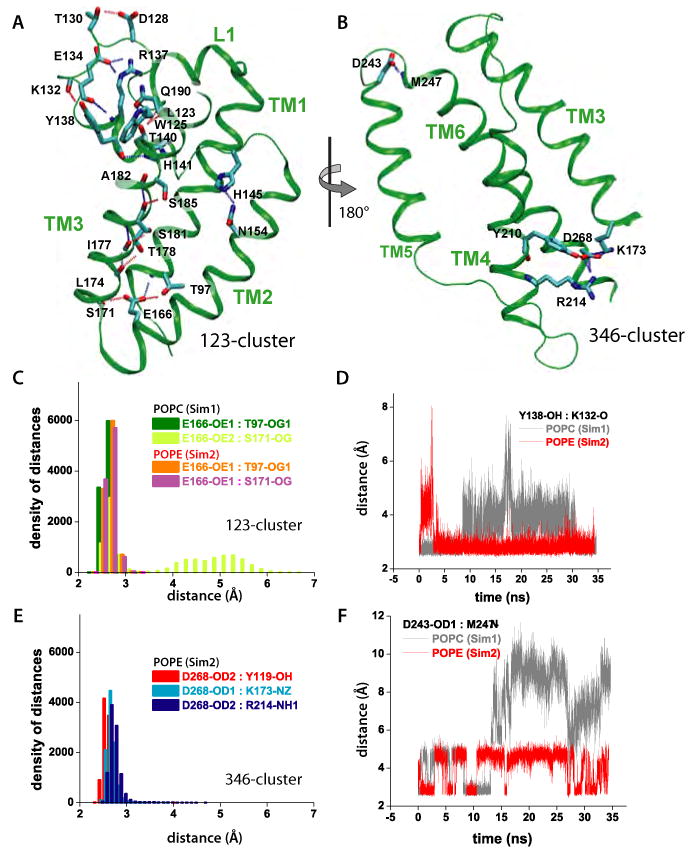
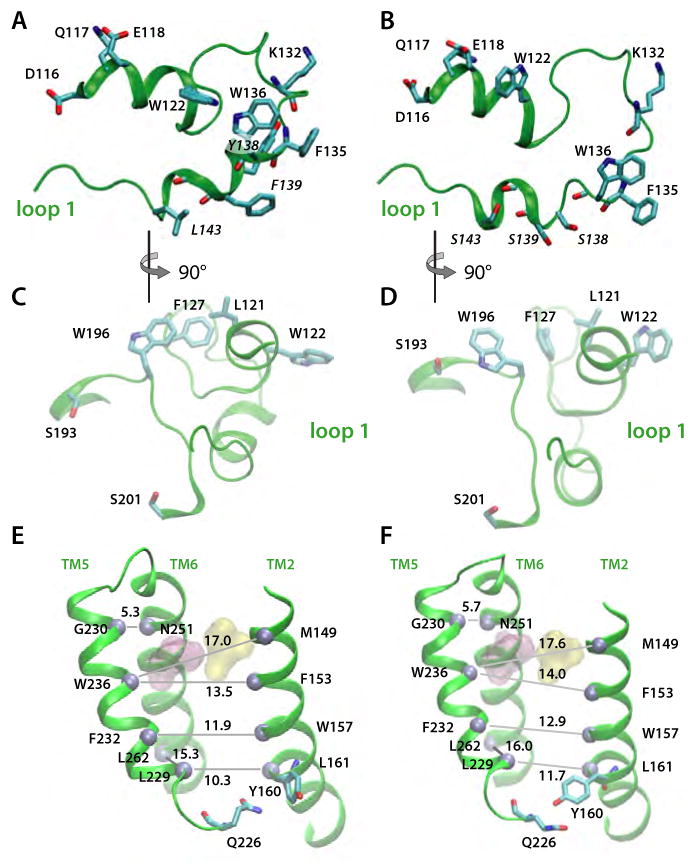
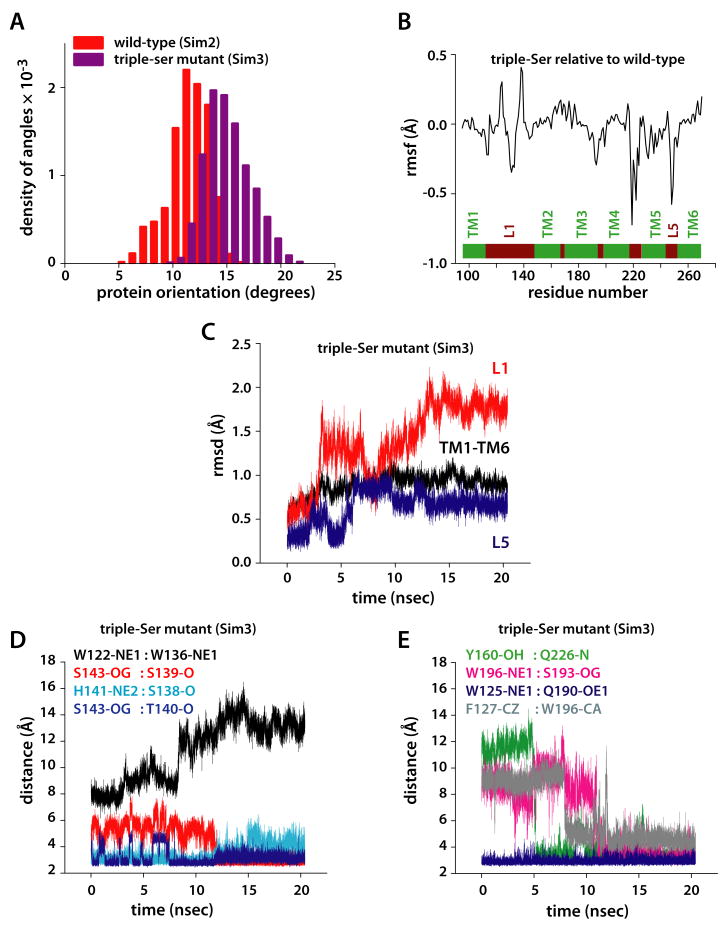
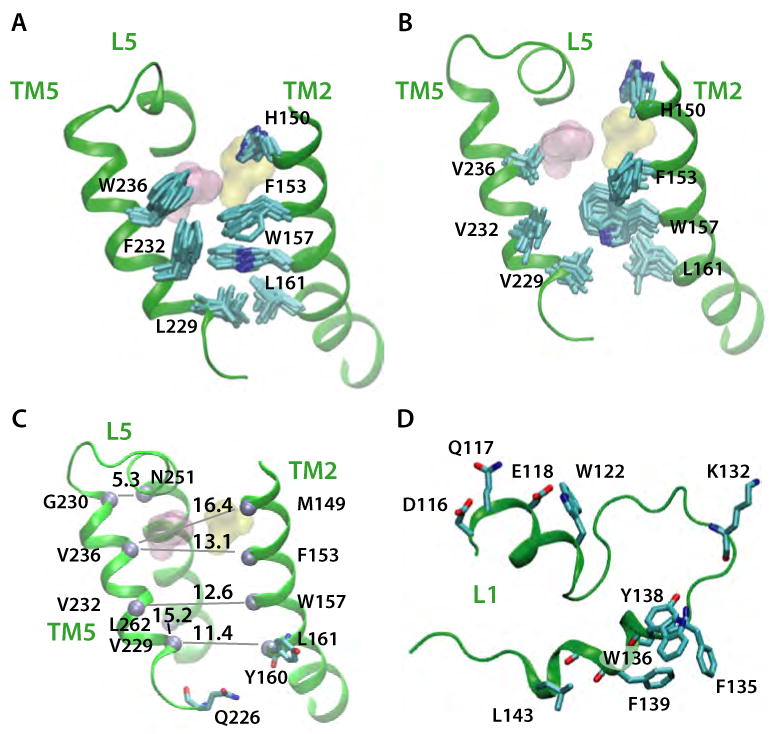
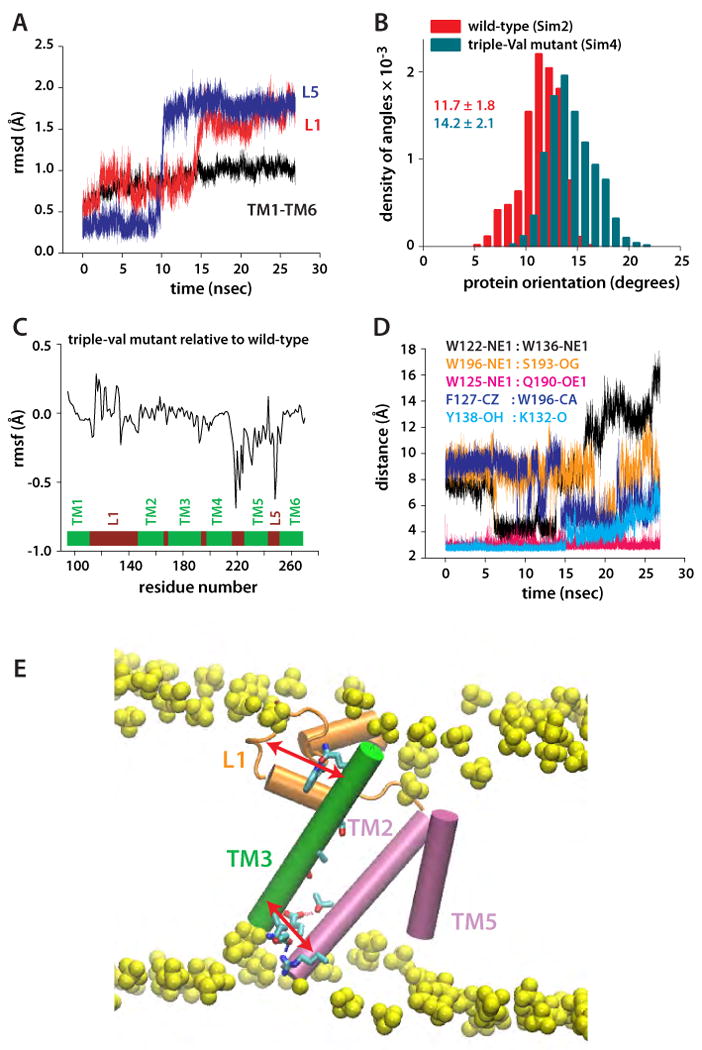
Intramembrane proteases, which cleave transmembrane (TM) helices, participate in numerous biological processes encompassing all branches of life. Several crystallographic structures of Escherichia coli GlpG rhomboid protease have been determined. In order to understand GlpG dynamics and lipid interactions in a native-like environment, we have examined the molecular dynamics of wild-type and mutant GlpG in different membrane environments. The irregular shape and small hydrophobic thickness of the protein cause significant bilayer deformations that may be important for substrate entry into the active site. Hydrogen-bond interactions with lipids are paramount in protein orientation and dynamics. Mutations in the unusual L1 loop cause changes in protein dynamics and protein orientation that are relayed to the His-Ser catalytic dyad. Similarly, mutations in TM5 change the dynamics and structure of the L1 loop. These results imply that the L1 loop has an important regulatory role in proteolysis.
Figures
References
-
- Baran KL, Chimenti MS, Schlessman JL, Fitch CA, Herbst KJ, Garcia-Moreno BE. Electrostatic effects in a network of polar and ionizable groups in Staphylococcal nuclease. J Mol Biol. 2008;379:1045–1062. - PubMed
-
- Bier E, Jan LY, Jan YN. rhomboid, a gene required for dosoventral axis establishment and peripheral nervous system develompent in Drosophila melanogaster. Genes Dev. 1990;4:190–203. - PubMed
-
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics. J Comput Chem. 1983;4:187–217.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
