

A tumor suppressor function for caspase-2
- PMID: 19279217
- PMCID: PMC2664004
- DOI: 10.1073/pnas.0811928106
A tumor suppressor function for caspase-2
Abstract
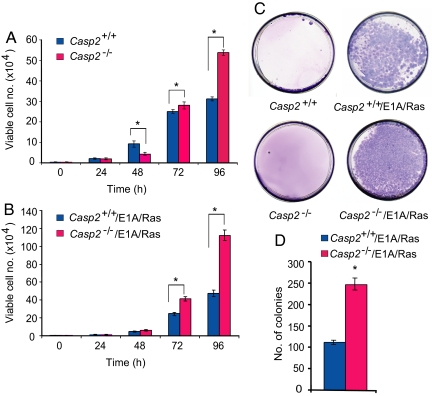
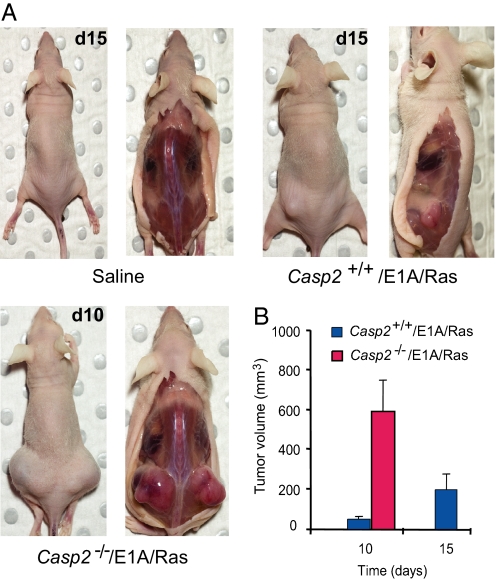
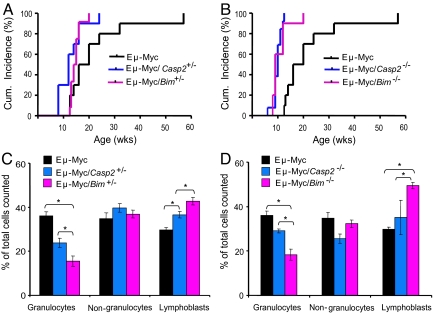
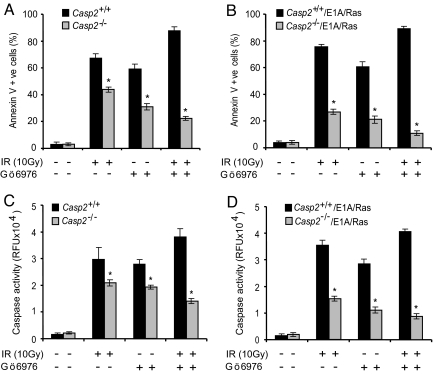
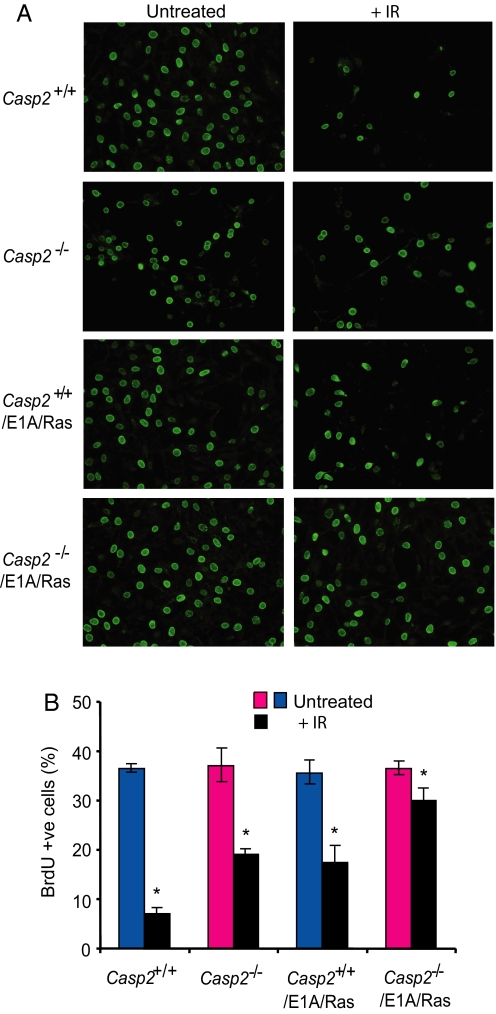
Apoptosis is mediated by the caspase family of proteases that act as effectors of cell death by cleaving many cellular substrates. Caspase-2 is one of the most evolutionarily conserved caspases, yet its physiological function has remained enigmatic because caspase-2-deficient mice develop normally and are viable. We report here that the caspase-2(-/-) mouse embryonic fibroblasts (MEFs) show increased proliferation. When transformed with E1A and Ras oncogenes, caspase-2(-/-) MEFs grew significantly faster than caspase-2(+/+) MEFs and formed more aggressive and accelerated tumors in nude mice. To assess whether the loss of caspase-2 predisposes animals to tumor development, we used the mouse Emu-Myc lymphoma model. Our findings suggest that loss of even a single allele of caspase-2 resulted in accelerated tumorigenesis, and this was further enhanced in caspase-2(-/-) mice. The caspase-2(-/-) cells showed resistance to apoptosis induced by chemotherapeutic drugs and DNA damage. Furthermore, caspase-2(-/-) MEFs had a defective apoptotic response to cell-cycle checkpoint regulation and showed abnormal cycling following gamma-irradiation. These data show that loss of caspase-2 results in an increased ability of cells to acquire a transformed phenotype and become malignant, indicating that caspase-2 is a tumor suppressor protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. - PubMed
-
- Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. - PubMed
-
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. - PubMed
-
- Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994;8:1613–1626. - PubMed
-
- Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
