

Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro
- PMID: 19286953
- PMCID: PMC2685360
- DOI: 10.1152/ajpheart.00795.2008
Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro
Abstract
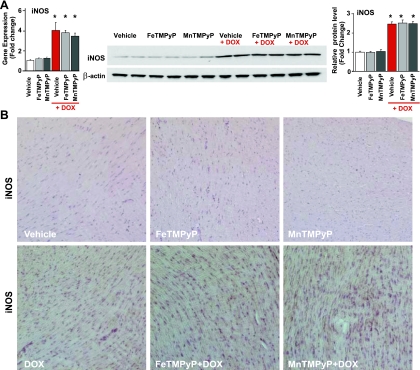
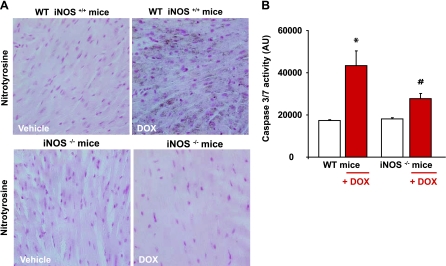
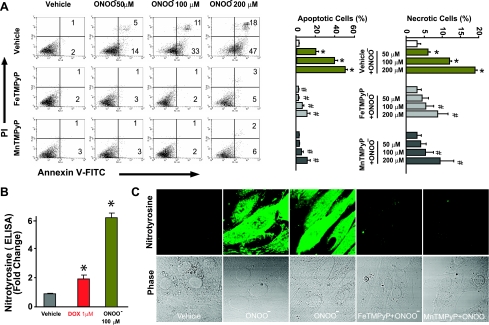
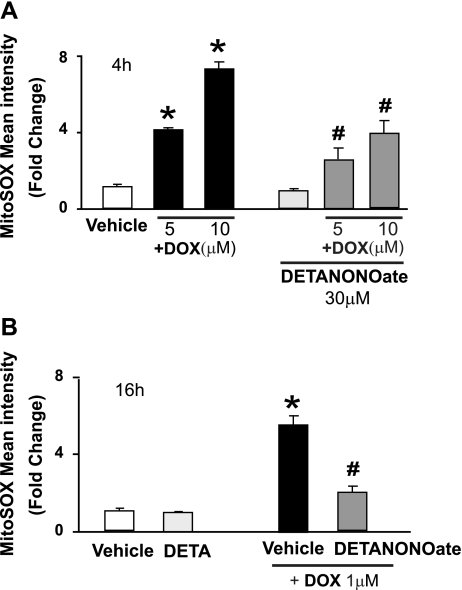
Doxorubicin (DOX) is a potent available antitumor agent; however, its clinical use is limited because of its cardiotoxicity. Cell death is a key component in DOX-induced cardiotoxicity, but its mechanisms are elusive. Here, we explore the role of superoxide, nitric oxide (NO), and peroxynitrite in DOX-induced cell death using both in vivo and in vitro models of cardiotoxicity. Western blot analysis, real-time PCR, immunohistochemistry, flow cytometry, fluorescent microscopy, and biochemical assays were used to determine the markers of apoptosis/necrosis and sources of NO and superoxide and their production. Left ventricular function was measured by a pressure-volume system. We demonstrated increases in myocardial apoptosis (caspase-3 cleavage/activity, cytochrome c release, and TUNEL), inducible NO synthase (iNOS) expression, mitochondrial superoxide generation, 3-nitrotyrosine (NT) formation, matrix metalloproteinase (MMP)-2/MMP-9 gene expression, poly(ADP-ribose) polymerase activation [without major changes in NAD(P)H oxidase isoform 1, NAD(P)H oxidase isoform 2, p22(phox), p40(phox), p47(phox), p67(phox), xanthine oxidase, endothelial NOS, and neuronal NOS expression] and decreases in myocardial contractility, catalase, and glutathione peroxidase activities 5 days after DOX treatment to mice. All these effects of DOX were markedly attenuated by peroxynitrite scavengers. Doxorubicin dose dependently increased mitochondrial superoxide and NT generation and apoptosis/necrosis in cardiac-derived H9c2 cells. DOX- or peroxynitrite-induced apoptosis/necrosis positively correlated with intracellular NT formation and could be abolished by peroxynitrite scavengers. DOX-induced cell death and NT formation were also attenuated by selective iNOS inhibitors or in iNOS knockout mice. Various NO donors when coadministered with DOX but not alone dramatically enhanced DOX-induced cell death with concomitant increased NT formation. DOX-induced cell death was also attenuated by cell-permeable SOD but not by cell-permeable catalase, the xanthine oxidase inhibitor allopurinol, or the NADPH oxidase inhibitors apocynine or diphenylene iodonium. Thus, peroxynitrite is a major trigger of DOX-induced cell death both in vivo and in vivo, and the modulation of the pathways leading to its generation or its effective neutralization can be of significant therapeutic benefit.
Figures











References
-
- Altmann SM, Muryshev A, Fossale E, Maxwell MM, Norflus FN, Fox J, Hersch SM, Young AB, MacDonald ME, Abagyan R, Kazantsev AG. Discovery of bioactive small-molecule inhibitor of poly ADP-ribose polymerase: implications for energy-deficient cells. Chem Biol 13: 765–770, 2006. - PubMed
-
- Andreadou I, Sigala F, Iliodromitis EK, Papaefthimiou M, Sigalas C, Aligiannis N, Savvari P, Gorgoulis V, Papalabros E, Kremastinos DT. Acute doxorubicin cardiotoxicity is successfully treated with the phytochemical oleuropein through suppression of oxidative and nitrosative stress. J Mol Cell Cardiol 42: 549–558, 2007. - PubMed
-
- Bai P, Mabley JG, Liaudet L, Virág L, Szabó C, Pacher P. Matrix metalloproteinase activation is an early event in doxorubicin-induced cardiotoxicity. Oncol Rep 11: 505–508, 2004. - PubMed
-
- Bátkai S, Rajesh M, Mukhopadhyay P, Haskó G, Liaudet L, Cravatt BF, Csiszár A, Ungvári Z, Pacher P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am J Physiol Heart Circ Physiol 293: H909–H918, 2007. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials