

Human copy number polymorphic genes
- PMID: 19287160
- PMCID: PMC2920189
- DOI: 10.1159/000184713
Human copy number polymorphic genes
Abstract
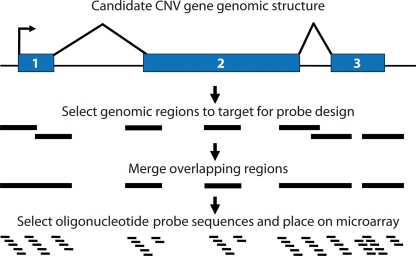
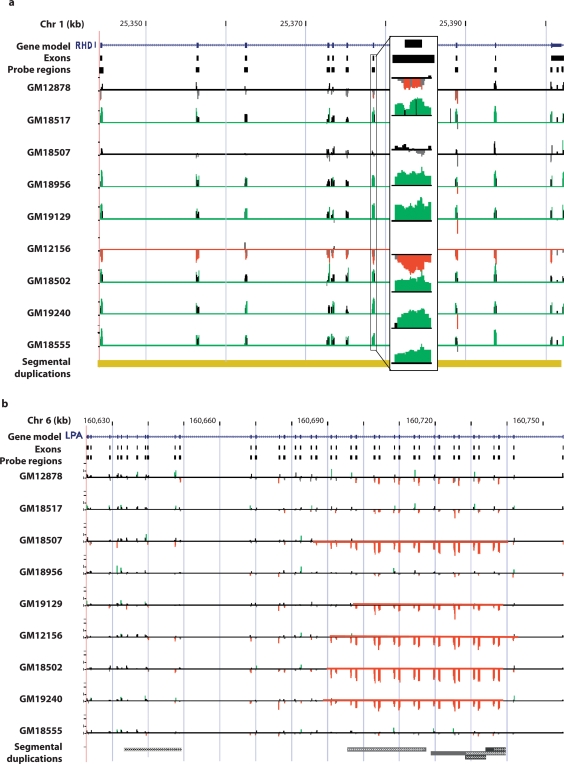
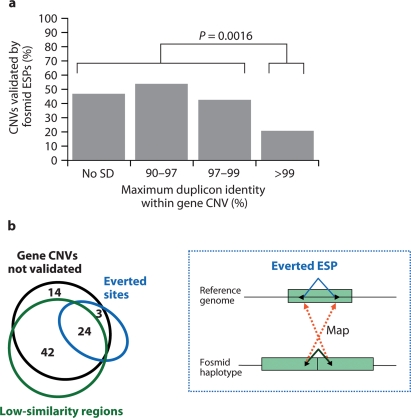
Recent large-scale genomic studies within human populations have identified numerous genomic regions as copy number variant (CNV). As these CNV regions often overlap coding regions of the genome, large lists of potentially copy number polymorphic genes have been produced that are candidates for disease association. Most of the current data regarding normal genic variation, however, has been generated using BAC or SNP microarrays, which lack precision especially with respect to exons. To address this, we assessed 2,790 candidate CNV genes defined from available studies in nine well-characterized HapMap individuals by designing a customized oligonucleotide microarray targeted specifically to exons. Using exon array comparative genomic hybridization (aCGH), we detected 255 (9%) of the candidates as true CNVs including 134 with evidence of variation over the entire gene. Individuals differed in copy number from the control by an average of 100 gene loci. Both partial- and whole-gene CNVs were strongly associated with segmental duplications (55 and 71%, respectively) as well as regions of positive selection. We confirmed 37% of the whole-gene CNVs using the fosmid end sequence pair (ESP) structural variation map for these same individuals. If we modify the end sequence pair mapping strategy to include low-sequence identity ESPs (98-99.5%) and ESPs with an everted orientation, we can capture 82% of the missed genes leading to more complete ascertainment of structural variation within duplicated genes. Our results indicate that segmental duplications are the source of the majority of full-length copy number polymorphic genes, most of the variant genes are organized as tandem duplications, and a significant fraction of these genes will represent paralogs with levels of sequence diversity beyond thresholds of allelic variation. In addition, these data provide a targeted set of CNV genes enriched for regions likely to be associated with human phenotypic differences due to copy number changes and present a source of copy number responsive oligonucleotide probes for future association studies.
Copyright 2009 S. Karger AG, Basel.
Figures
References
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. - PubMed
-
- Barber JC, Reed CJ, Dahoun SP, Joyce CA. Amplification of a pseudogene cassette underlies euchromatic variation of 16p at the cytogenetic level. Hum Genet. 1999;104:211–218. - PubMed
-
- Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. - PubMed
-
- Benjamini Y, Hochberg Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
