

FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation
- PMID: 19287496
- PMCID: PMC2654098
- DOI: 10.1371/journal.pone.0004849
FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation
Abstract
Background: Cancer associated with smoking and drinking remains a serious health problem worldwide. The survival of patients is very poor due to the lack of effective early biomarkers. FOXM1 overexpression is linked to the majority of human cancers but its mechanism remains unclear in head and neck squamous cell carcinoma (HNSCC).
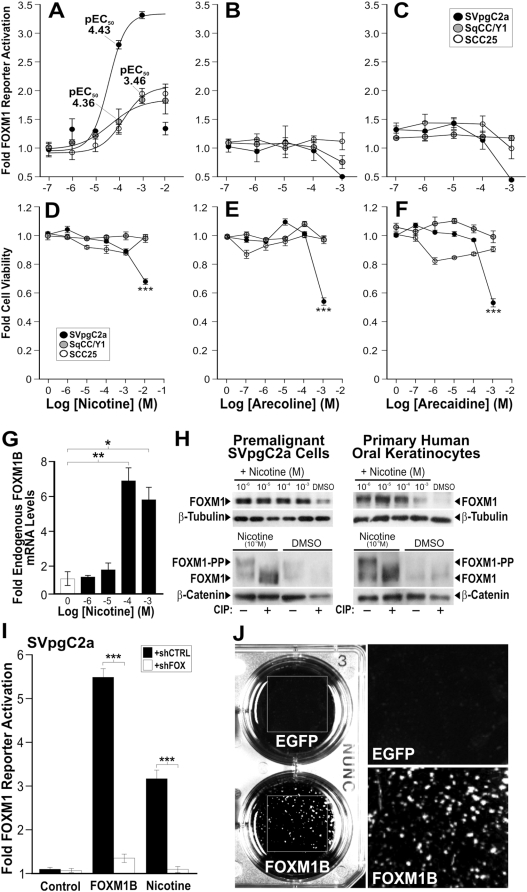
Methodology/principal findings: FOXM1 mRNA and protein expressions were investigated in four independent cohorts (total 75 patients) consisting of normal, premalignant and HNSCC tissues and cells using quantitative PCR (qPCR), expression microarray, immunohistochemistry and immunocytochemistry. Effect of putative oral carcinogens on FOXM1 transcriptional activity was dose-dependently assayed and confirmed using a FOXM1-specific luciferase reporter system, qPCR, immunoblotting and short-hairpin RNA interference. Genome-wide single nucleotide polymorphism (SNP) array was used to 'trace' the genomic instability signature pattern in 8 clonal lines of FOXM1-induced malignant human oral keratinocytes. Furthermore, acute FOXM1 upregulation in primary oral keratinocytes directly induced genomic instability. We have shown for the first time that overexpression of FOXM1 precedes HNSCC malignancy. Screening putative carcinogens in human oral keratinocytes surprisingly showed that nicotine, which is not perceived to be a human carcinogen, directly induced FOXM1 mRNA, protein stabilisation and transcriptional activity at concentrations relevant to tobacco chewers. Importantly, nicotine also augmented FOXM1-induced transformation of human oral keratinocytes. A centrosomal protein CEP55 and a DNA helicase/putative stem cell marker HELLS, both located within a consensus loci (10q23), were found to be novel targets of FOXM1 and their expression correlated tightly with HNSCC progression.
Conclusions/significance: This study cautions the potential co-carcinogenic effect of nicotine in tobacco replacement therapies. We hypothesise that aberrant upregulation of FOXM1 may be inducing genomic instability through a program of malignant transformation involving the activation of CEP55 and HELLS which may facilitate aberrant mitosis and epigenetic modifications. Our finding that FOXM1 is upregulated early during oral cancer progression renders FOXM1 an attractive diagnostic biomarker for early cancer detection and its candidate mechanistic targets, CEP55 and HELLS, as indicators of malignant conversion and progression.
Conflict of interest statement
Figures







References
-
- Katoh M. Human FOX gene family (Review). Int J Oncol. 2004;25:1495–1500. - PubMed
-
- Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257–1274. - PubMed
-
- Korver W, Schilham MW, Moerer P, van den Hoff MJ, Dam K, et al. Uncoupling of S phase and mitosis in cardiomyocytes and hepatocytes lacking the winged-helix transcription factor Trident. Curr Biol. 1998;8:1327–1330. - PubMed
-
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. - PubMed
-
- Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, et al. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
