

Energetic profiling of protein folds
- PMID: 19289211
- PMCID: PMC5823608
- DOI: 10.1016/S0076-6879(08)04211-0
Energetic profiling of protein folds
Abstract
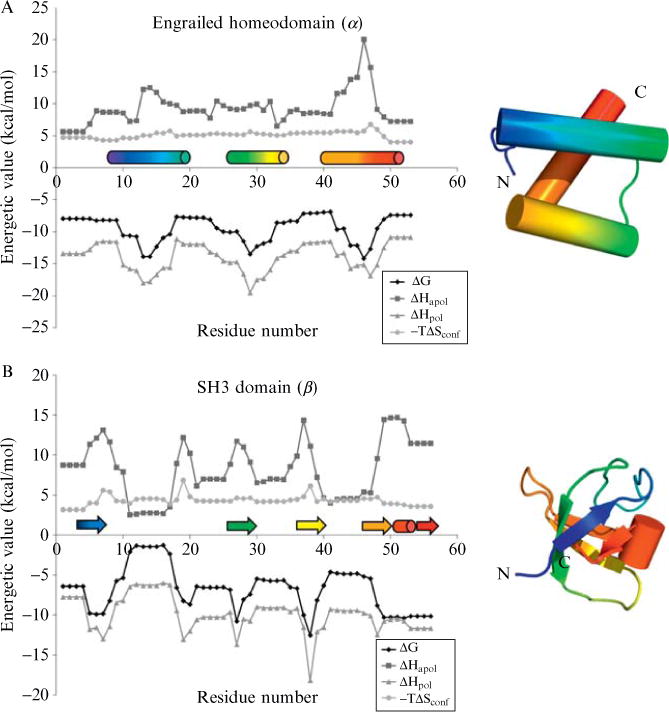
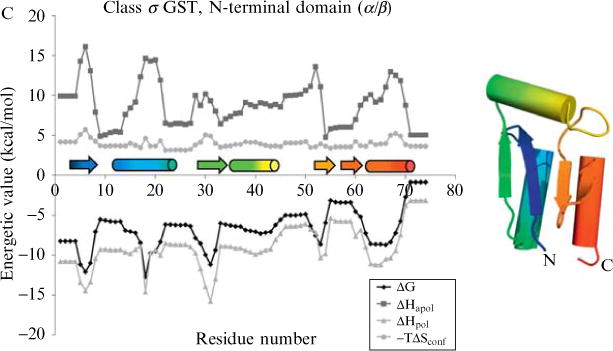
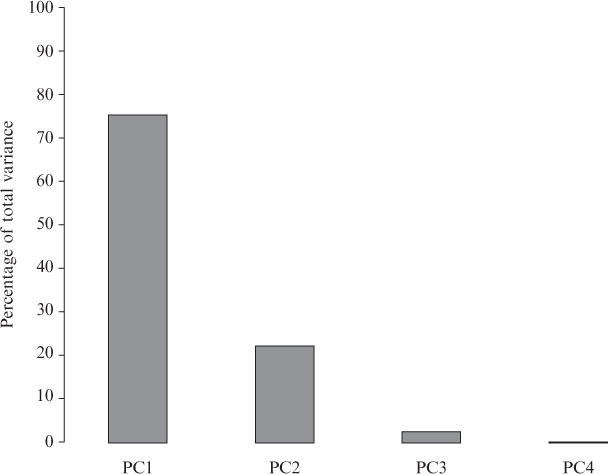
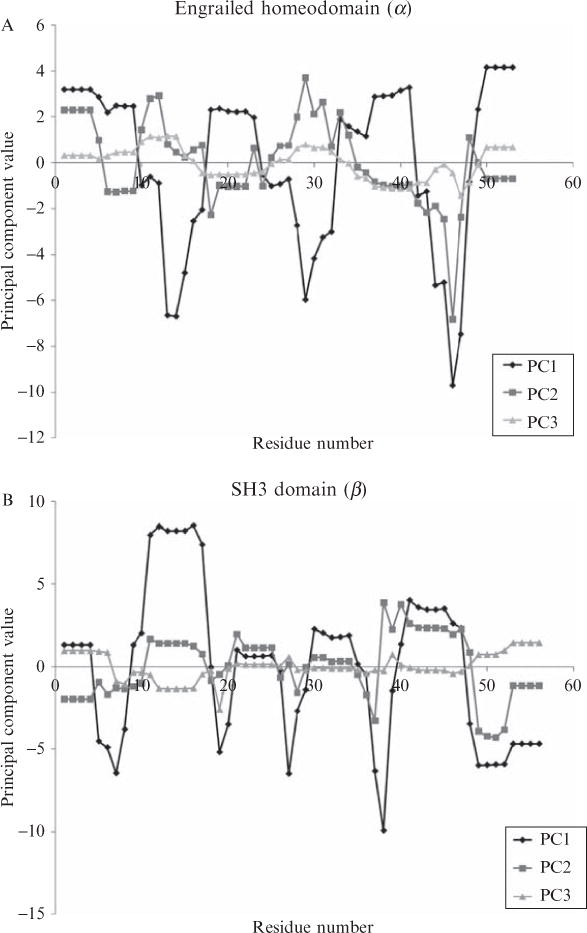
Current protein classification methods treat high-resolution structures as static entities. However, experiments have well documented the dynamic nature of proteins. With knowledge that thermodynamic fluctuations around the high-resolution structure contribute to a more physically accurate and biologically meaningful picture of a protein, the concept of a protein's energetic profile is introduced. It is demonstrated on a large scale that energetic profiles are both diagnostic of a protein fold and evolutionarily relevant. Development of Structural Thermodynamic Ensemble-based Protein Homology (STEPH), an algorithm that searches for local similarities between energetic profiles, constitutes a first step towards a long-term goal of our laboratory to integrate thermodynamic information into protein-fold classification approaches.
Figures

Similar articles
-
Thermodynamic environments in proteins: fundamental determinants of fold specificity.Protein Sci. 2002 Aug;11(8):1945-57. doi: 10.1110/ps.0203202. Protein Sci. 2002. PMID: 12142449 Free PMC article.
-
Denatured-state energy landscapes of a protein structural database reveal the energetic determinants of a framework model for folding.J Mol Biol. 2008 Sep 19;381(5):1184-201. doi: 10.1016/j.jmb.2008.06.046. Epub 2008 Jun 24. J Mol Biol. 2008. PMID: 18616947 Free PMC article.
-
Analysis of the "thermodynamic information content" of a Homo sapiens structural database reveals hierarchical thermodynamic organization.Protein Sci. 2004 Jul;13(7):1787-801. doi: 10.1110/ps.04706204. Protein Sci. 2004. PMID: 15215522 Free PMC article.
-
Fluctuations within folded proteins: implications for thermodynamic and allosteric regulation.Acc Chem Res. 2015 Apr 21;48(4):1098-105. doi: 10.1021/ar500351b. Epub 2015 Feb 17. Acc Chem Res. 2015. PMID: 25688669 Review.
-
Acquisition and use of calorimetric data for prediction of the thermodynamics of ligand-binding and folding reactions of proteins.Curr Opin Biotechnol. 1994 Aug;5(4):381-8. doi: 10.1016/0958-1669(94)90046-9. Curr Opin Biotechnol. 1994. PMID: 7765170 Review.
Cited by
-
The role of protein conformational fluctuations in allostery, function, and evolution.Biophys Chem. 2011 Nov;159(1):129-41. doi: 10.1016/j.bpc.2011.05.020. Epub 2011 May 31. Biophys Chem. 2011. PMID: 21684672 Free PMC article. Review.
-
The role of key residues in structure, function, and stability of cytochrome-c.Cell Mol Life Sci. 2014 Jan;71(2):229-55. doi: 10.1007/s00018-013-1341-1. Epub 2013 Apr 25. Cell Mol Life Sci. 2014. PMID: 23615770 Free PMC article. Review.
-
Quantitative assessment of protein structural models by comparison of H/D exchange MS data with exchange behavior accurately predicted by DXCOREX.J Am Soc Mass Spectrom. 2012 Jan;23(1):43-56. doi: 10.1007/s13361-011-0267-9. Epub 2011 Oct 20. J Am Soc Mass Spectrom. 2012. PMID: 22012689 Free PMC article.
-
Protein hydrogen exchange: testing current models.Protein Sci. 2012 Jul;21(7):987-95. doi: 10.1002/pro.2082. Epub 2012 Jun 11. Protein Sci. 2012. PMID: 22544567 Free PMC article.
-
Prediction of protein relative enthalpic stability from molecular dynamics simulations of the folded and unfolded states.Biophys J. 2012 Oct 17;103(8):1762-73. doi: 10.1016/j.bpj.2012.08.048. Epub 2012 Oct 16. Biophys J. 2012. PMID: 23083720 Free PMC article.
References
-
- Alva V, Koretke KK, Coles M, Lupas AN. Cradle-loop barrels and the concept of metafolds in protein classification by natural descent. Curr Opin Struct Biol. 2008;18:358–365. - PubMed
-
- Babu CR, Hilser VJ, Wand AJ. Direct access to the cooperative substructure of proteins and the protein ensemble via cold denaturation. Nat Struct Mol Biol. 2004;11:352–357. - PubMed
-
- Chen L, Zhou T, Tang Y. Protein structure alignment by deterministic annealing. Bioinformatics. 2005;21:51–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
