

Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41
- PMID: 19289833
- PMCID: PMC2664029
- DOI: 10.1073/pnas.0811713106
Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41
Abstract
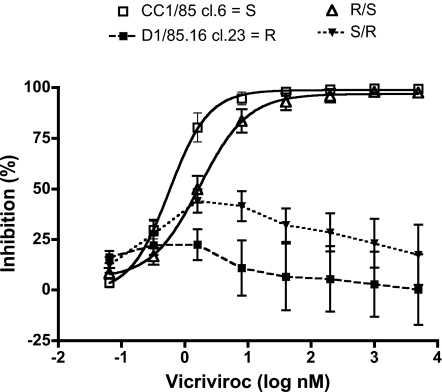
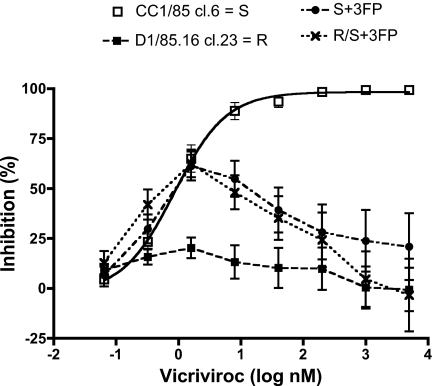
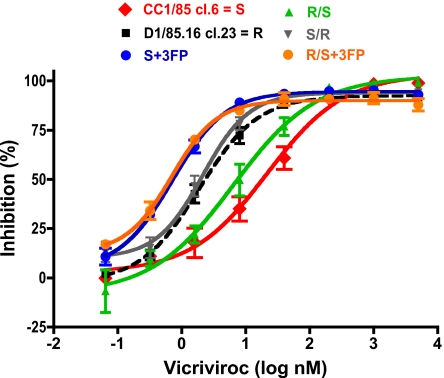
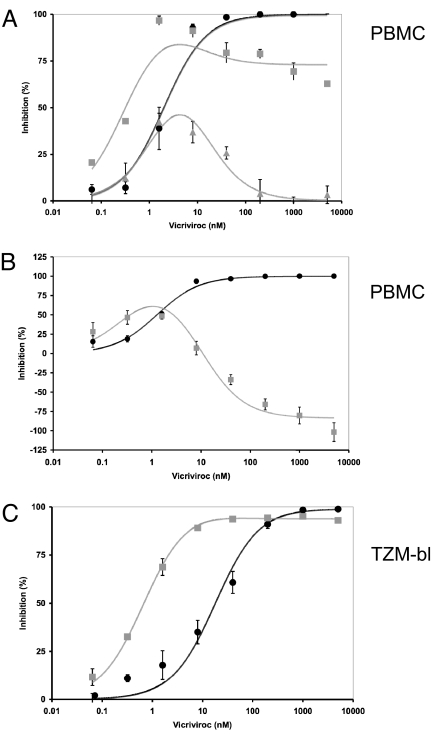
We have investigated the mechanism of resistance of a HIV type 1 (HIV-1) R5 primary isolate, D1/85.16, to the small molecule CCR5 inhibitor, vicriviroc (VVC). Unlike other viruses resistant to this class of compound, D1/85.16 lacks sequence changes in the V3 region of the gp120 surface glycoprotein. Inspection of env sequences from D1/85.16 compared with those derived from the parental, inhibitor-sensitive virus, CC1/85, revealed a cluster of 3 conservative changes in the fusion peptide (FP) of the gp41 transmembrane glycoprotein that tracked with the resistance phenotype. Studies with engineered Env-chimeric and point-substituted viruses confirmed that these 3 FP residues were substantially responsible for VVC resistance without altering coreceptor usage, as assessed in both peripheral blood mononuclear cells and the TZM-bl cell line. VVC resistance is manifested differently in the 2 cell types, and there are assay-dependent complexities to the dose-response curves for the engineered resistant viruses. To explain them, we created a model for resistance and generated theoretical VVC inhibition curves that closely mimic the experimental data for the resistant viruses. The basis for the model is the existence of distinct forms of CCR5, with varying affinities for small molecule CCR5 inhibitors that are presumed to be present in different proportions on different cell types, and are used selectively by resistant HIV-1 variants when ligated with an inhibitor. Together, the experimental results and theoretical model may help understand how HIV-1 uses CCR5 to enter target cells under various conditions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Kuhmann SE, Hartley O. Targeting chemokine receptors in HIV: A status report. Annu Rev Pharmacol Toxicol. 2008;48:425–461. - PubMed
-
- Moore JP, Kitchen SG, Pugach P, Zack JA. The CCR5 and CXCR4 coreceptors-central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 2004;20:111–126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
