

Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain
- PMID: 19293235
- PMCID: PMC2685919
- DOI: 10.1093/brain/awp049
Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain
Abstract
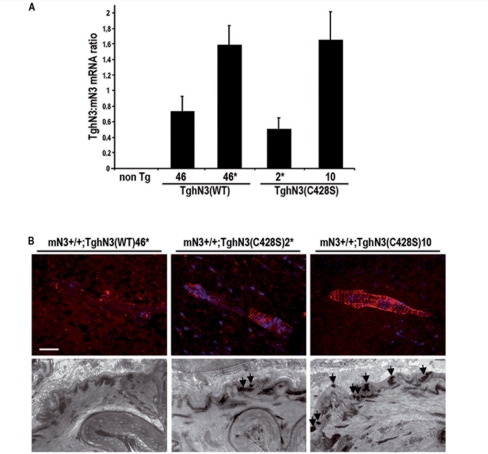
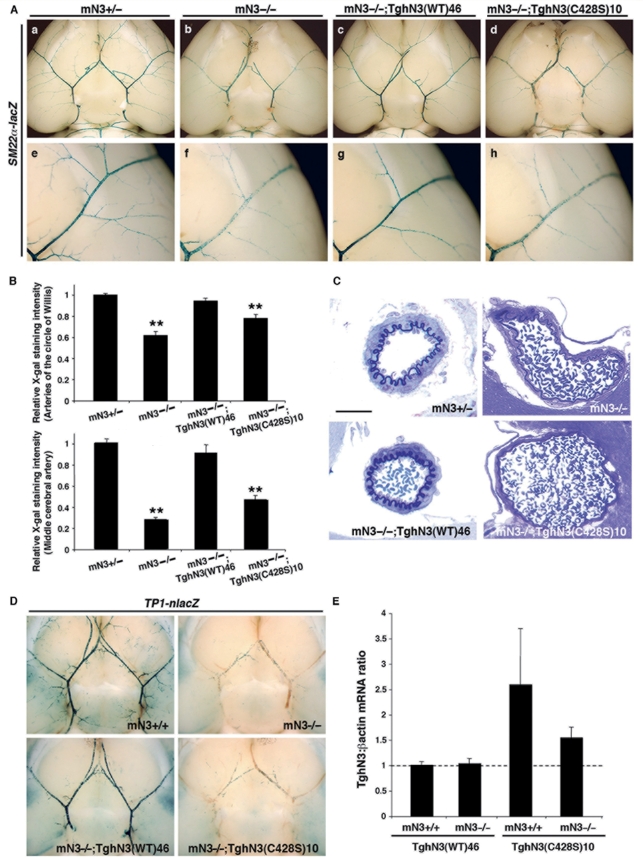
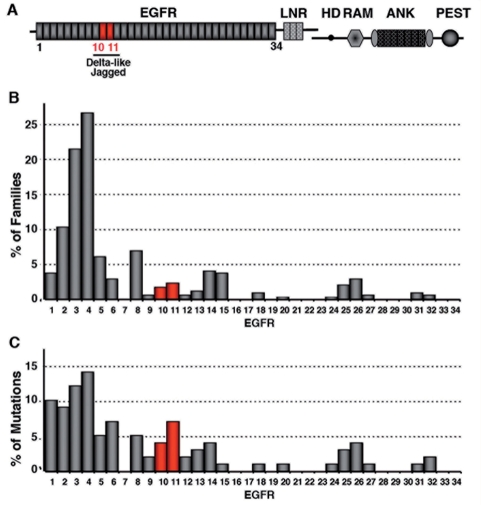
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autosomal dominant small-vessel disease of the brain caused by mutations in the NOTCH3 receptor. The highly stereotyped nature of the mutations, which alter the number of cysteine residues within the epidermal growth factor-like repeats (EGFR), predicts that all mutations share common mechanisms. Prior in vitro assays and genetic studies in the mouse support the hypothesis that common mutations do not compromise canonical Notch3 function but instead convey a non-physiological and deleterious activity to the receptor through the unpaired cysteine residue. Intriguingly, in vitro studies predict that mutations located in the Delta/Serrate/LAG-2 ligand binding domain-(EGFR10-11) may result in a loss of Notch3 receptor function. However, the in vivo relevance and functional significance of this with respect to the pathogenic mechanisms and clinical expression of the disease remain largely unexplored. To ascertain, in vivo, the functional significance of EGFR10-11 mutations, we generated transgenic mice with one representative mutation (C428S) in EGFR10 of Notch3. These mice, like those with a common R90C mutation, developed characteristic arterial accumulation of Notch3 protein and granular osmiophilic material upon aging. By introducing the mutant C428S transgene into a Notch3 null background, we found that, unlike the R90C mutant protein, the C428S mutant protein has lost wild-type Notch3 activity and exhibited mild dominant-negative activity in three different biological settings. From a large prospectively recruited cohort of 176 CADASIL patients, we identified 10 patients, from five distinct pedigrees carrying a mutation in EGFR10 or 11. These mutations were associated with significantly higher Mini-Mental State Examination and Mattis Dementia Rating Scale scores (P < 0.05), when compared with common mutations. Additionally, we found a strong effect of this genotype on the burden of white matter hyperintensities (P < 0.01). Collectively, these results highlight distinctive functional and phenotypic features of EGFR10-11 mutations relative to the common CADASIL mutations. Our findings are compatible with the hypothesis that EGFR10-11 mutations cause the disease through the same gain of novel function as the common mutations, and lead us to propose that reduced Notch3 signalling acts as a modifier of the CADASIL phenotype.
Figures

References
-
- Arboleda-Velasquez JF, Lopera F, Lopez E, Frosch MP, Sepulveda-Falla D, Gutierrez JE, et al. C455R notch3 mutation in a Colombian CADASIL kindred with early onset of stroke. Neurology. 2002;59:277–9. - PubMed
-
- Chabriat H, Pappata S, Poupon C, Clark CA, Vahedi K, Poupon F, et al. Clinical severity in CADASIL related to ultrastructural damage in white matter: in vivo study with diffusion tensor MRI. Stroke. 1999;30:2637–43. - PubMed
-
- Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, Nagy TG, et al. Clinical spectrum of CADASIL: a study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet. 1995;346:934–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
