

Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice
- PMID: 19296651
- PMCID: PMC2746568
- DOI: 10.1021/jm801343r
Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice
Abstract
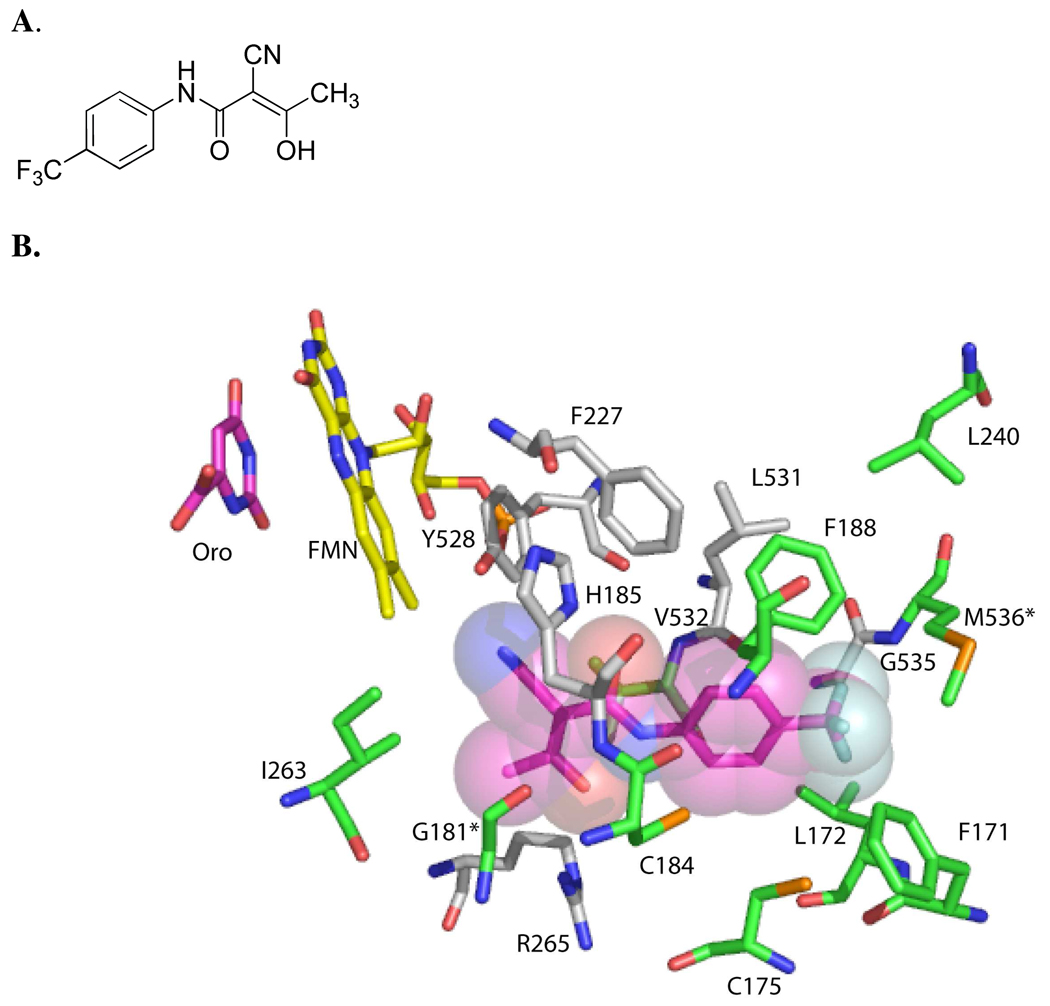
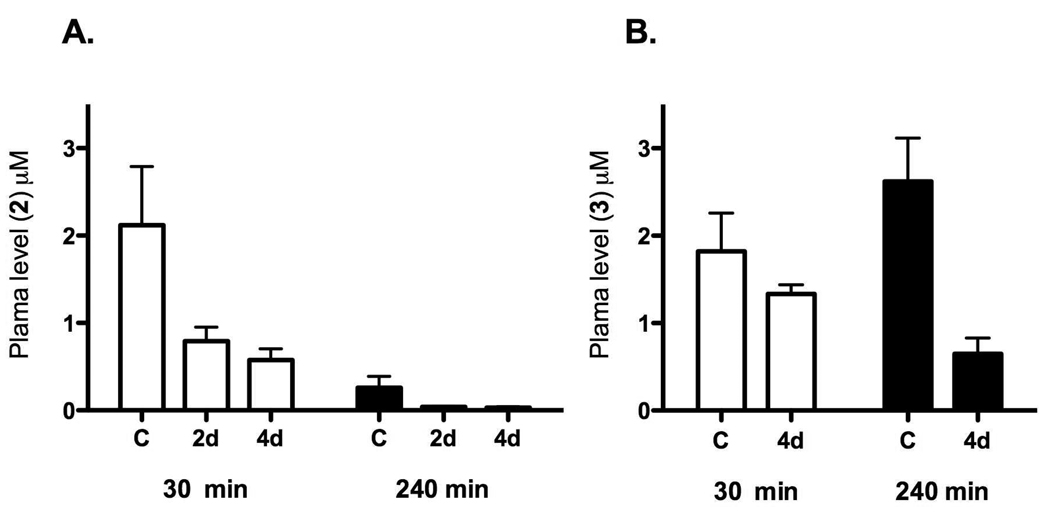
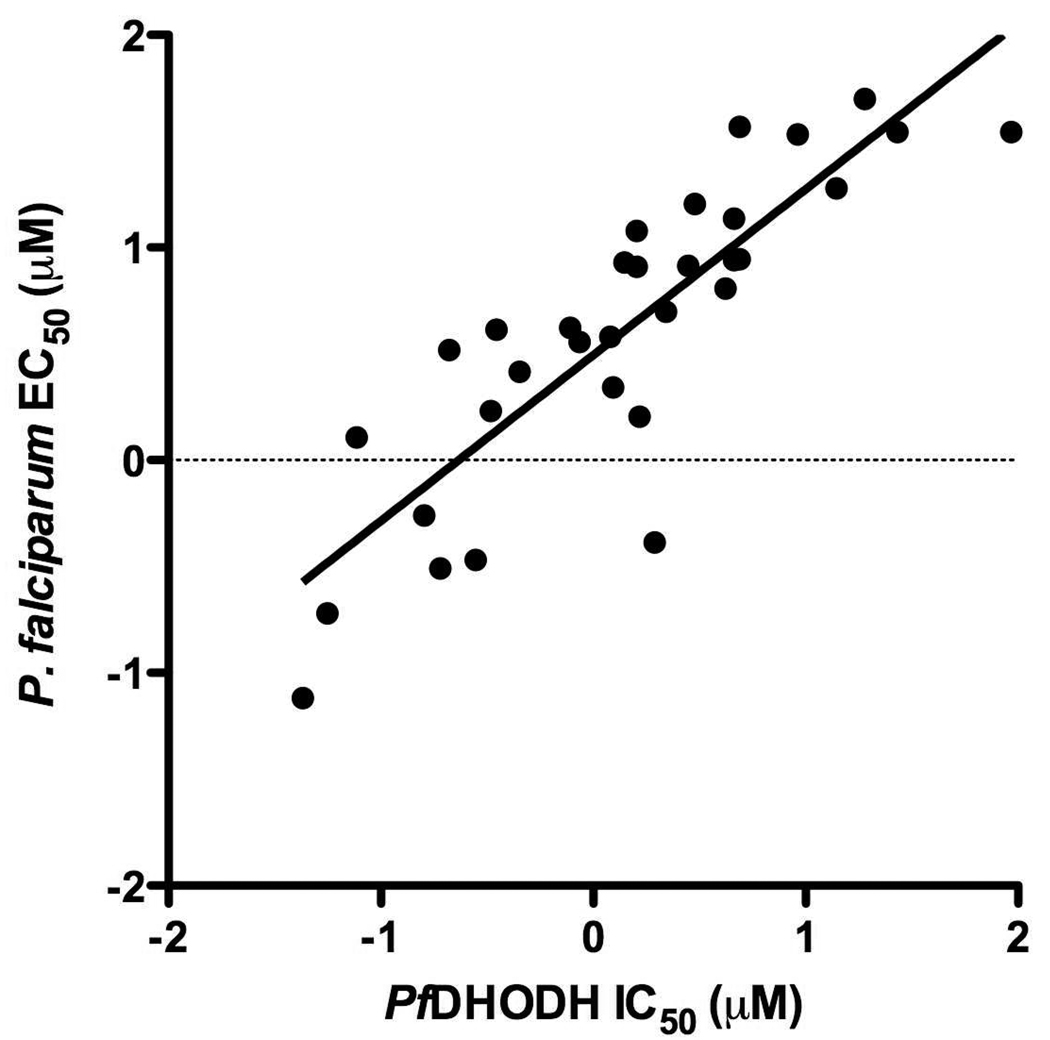
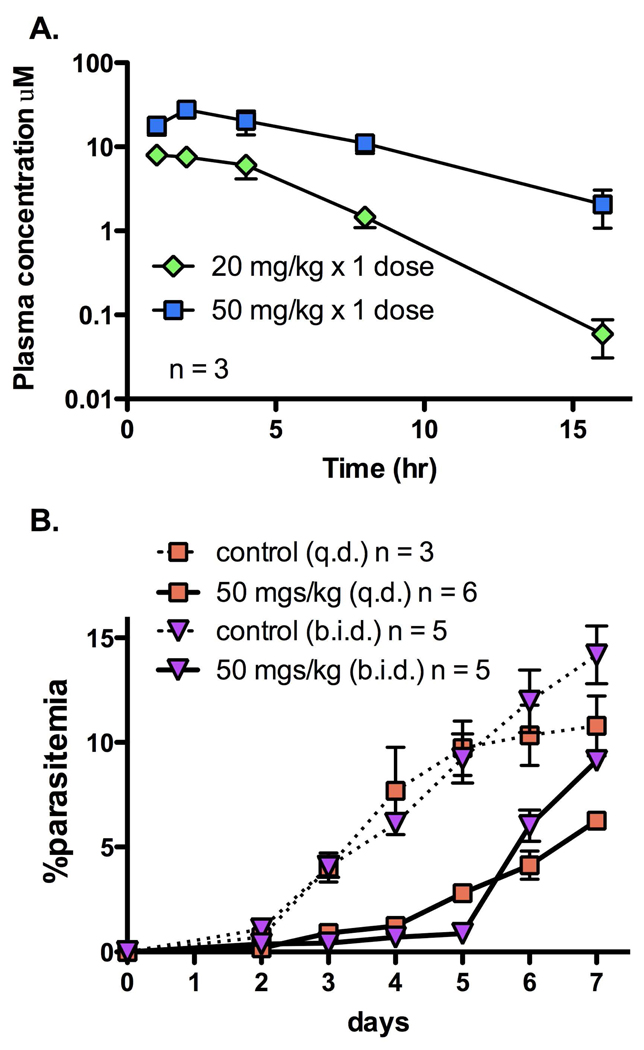
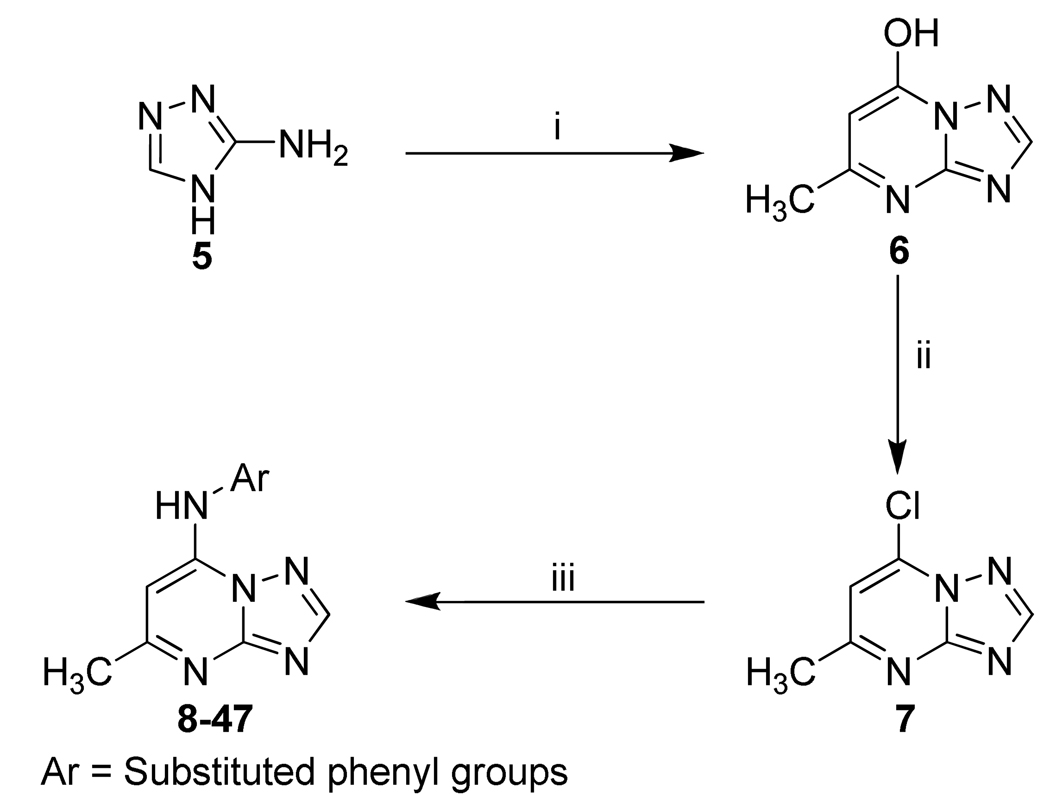
Plasmodium falciparum causes 1-2 million deaths annually. Yet current drug therapies are compromised by resistance. We previously described potent and selective triazolopyrimidine-based inhibitors of P. falciparum dihydroorotate dehydrogenase (PfDHODH) that inhibited parasite growth in vitro; however, they showed no activity in vivo. Here we show that lack of efficacy against P. berghei in mice resulted from a combination of poor plasma exposure and reduced potency against P. berghei DHODH. For compounds containing naphthyl (DSM1) or anthracenyl (DSM2), plasma exposure was reduced upon repeated dosing. Phenyl-substituted triazolopyrimidines were synthesized leading to identification of analogs with low predicted metabolism in human liver microsomes and which showed prolonged exposure in mice. Compound 21 (DSM74), containing p-trifluoromethylphenyl, suppressed growth of P. berghei in mice after oral administration. This study provides the first proof of concept that DHODH inhibitors can suppress Plasmodium growth in vivo, validating DHODH as a new target for antimalarial chemotherapy.
Figures
References
-
- Rosenthal PJ. Artesunate for the treatment of severe falciparum malaria. N Engl J Med. 2008;358:1829–1836. - PubMed
-
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Medical
