

Restricting activation-induced cytidine deaminase tumorigenic activity in B lymphocytes
- PMID: 19302140
- PMCID: PMC2669812
- DOI: 10.1111/j.1365-2567.2008.03050.x
Restricting activation-induced cytidine deaminase tumorigenic activity in B lymphocytes
Abstract
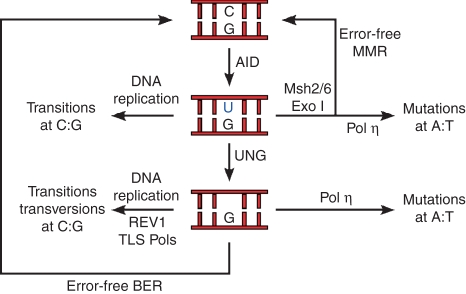
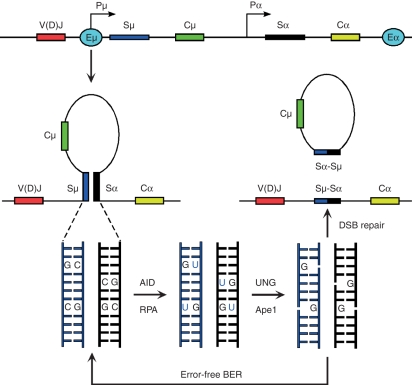
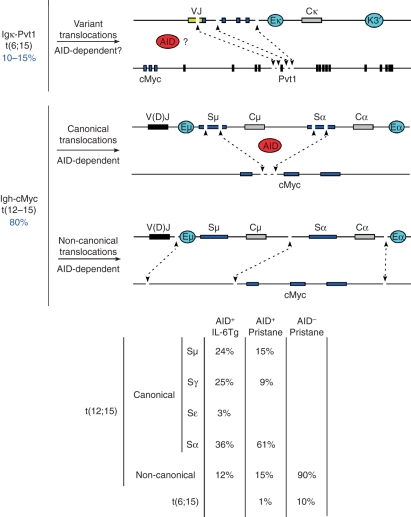
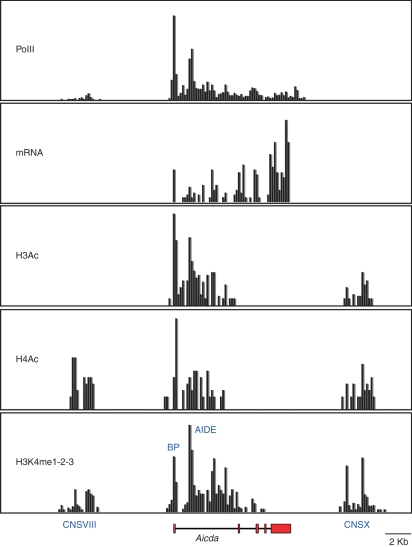
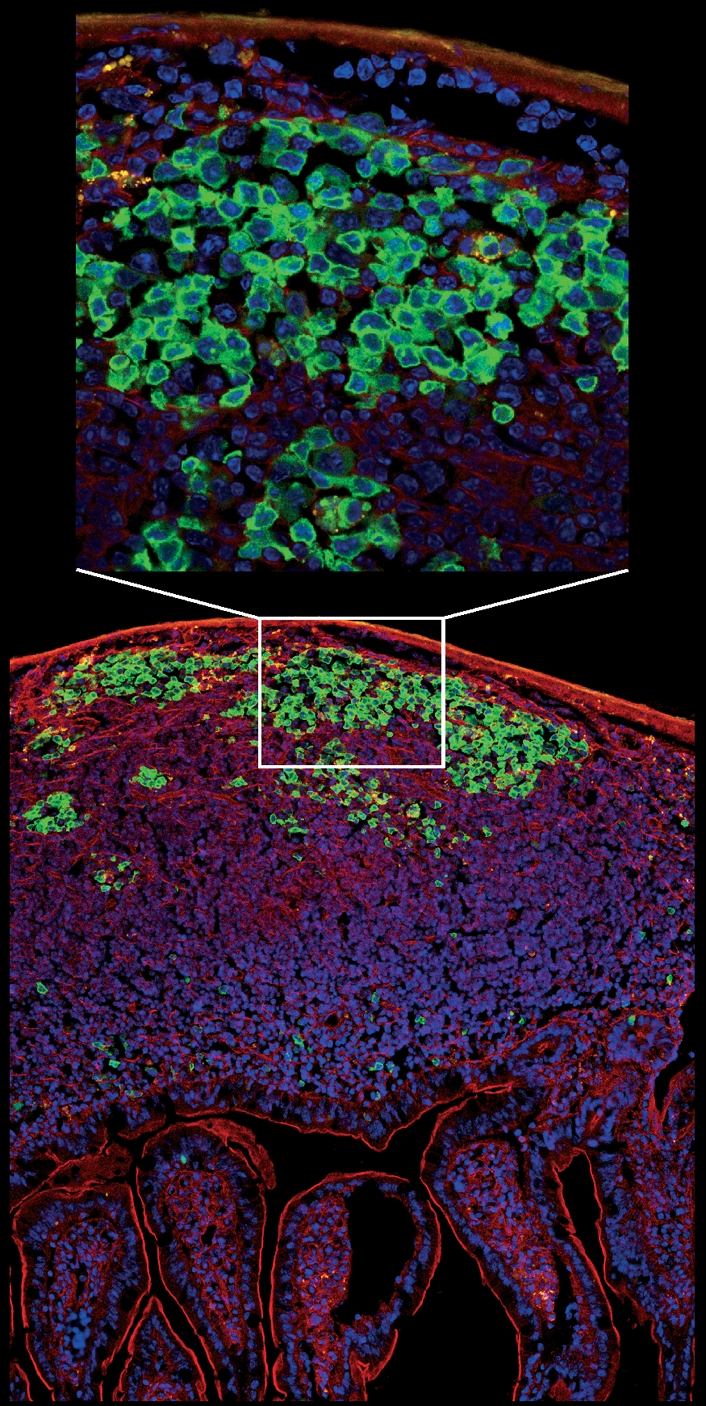
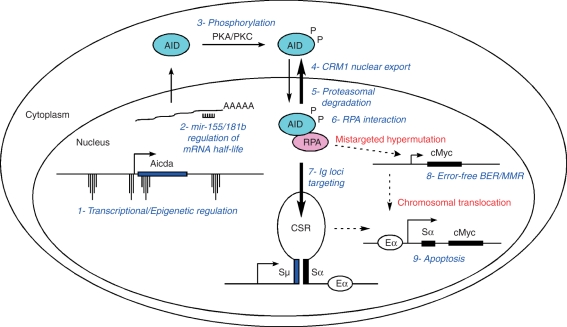
DNA breaks play an essential role in germinal centre B cells as intermediates to immunoglobulin class switching, a recombination process initiated by activation-induced cytidine deaminase (AID). Immunoglobulin gene hypermutation is likewise catalysed by AID but is believed to occur via single-strand DNA breaks. When improperly repaired, AID-mediated lesions can promote chromosomal translocations (CTs) that juxtapose the immunoglobulin loci to heterologous genomic sites, including oncogenes. Two of the most studied translocations are the t(8;14) and T(12;15), which deregulate cMyc in human Burkitt's lymphomas and mouse plasmacytomas, respectively. While a complete understanding of the aetiology of such translocations is lacking, recent studies using diverse mouse models have shed light on two important issues: (1) the extent to which non-specific or AID-mediated DNA lesions promote CTs, and (2) the safeguard mechanisms that B cells employ to prevent AID tumorigenic activity. Here we review these advances and discuss the usage of pristane-induced mouse plasmacytomas as a tool to investigate the origin of Igh-cMyc translocations and B-cell tumorigenesis.
Figures
References
-
- Neuberger MS. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–32. - PubMed
-
- Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002;20:165–96. - PubMed
-
- Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). [see comments] Cell. 2000;102:565–75. - PubMed
-
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. [see comments] Cell. 2000;102:553–63. - PubMed
-
- Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS. AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr Biol. 2002;12:435–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
