

Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin
- PMID: 19303373
- PMCID: PMC2693503
- DOI: 10.1016/j.dnarep.2009.02.002
Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin
Abstract
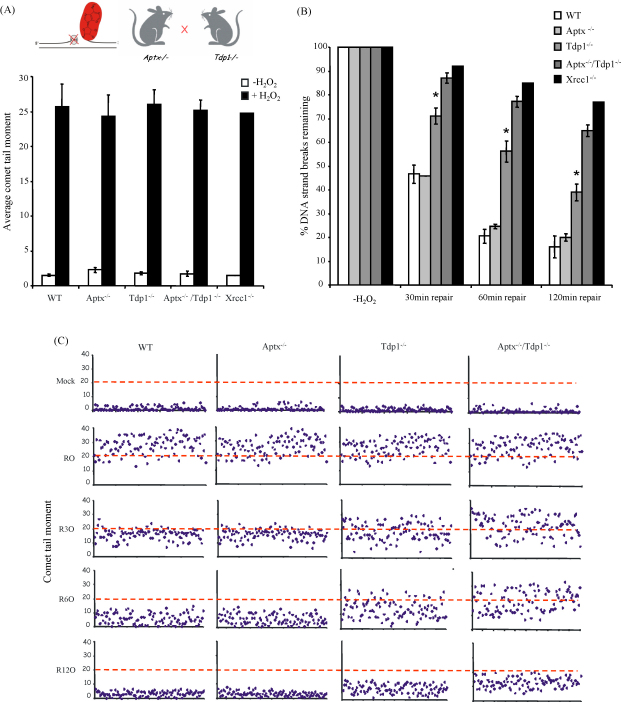
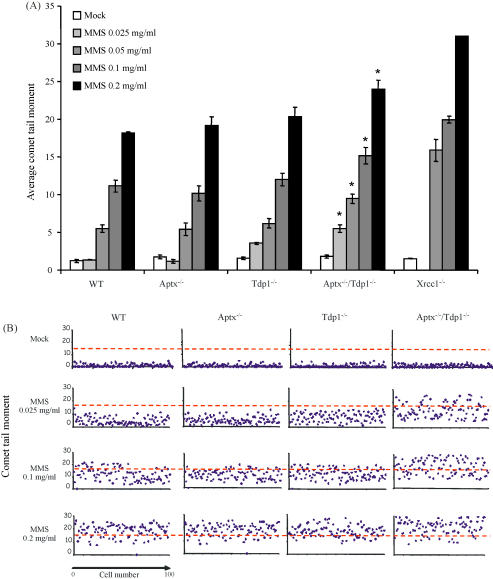
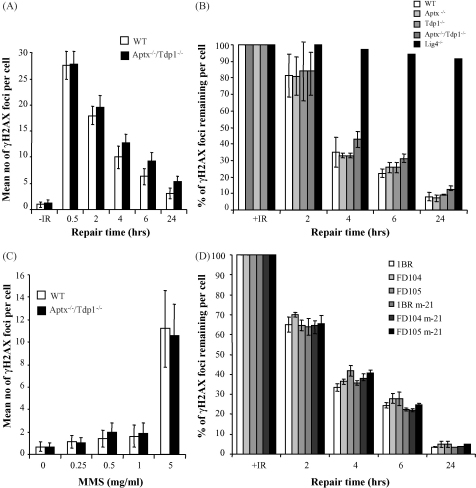
Ataxia oculomotor apraxia-1 (AOA1) is an autosomal recessive neurodegenerative disease that results from mutations of aprataxin (APTX). APTX associates with the DNA single- and double-strand break repair machinery and is able to remove AMP from 5'-termini at DNA strand breaks in vitro. However, attempts to establish a DNA strand break repair defect in APTX-defective cells have proved conflicting and unclear. We reasoned that this may reflect that DNA strand breaks with 5'-AMP represent only a minor subset of breaks induced in cells, and/or the availability of alternative mechanisms for removing AMP from 5'-termini. Here, we have attempted to increase the dependency of chromosomal single- and double-strand break repair on aprataxin activity by slowing the rate of repair of 3'-termini in aprataxin-defective neural cells, thereby increasing the likelihood that the 5'-termini at such breaks become adenylated and/or block alternative repair mechanisms. To do this, we generated a mouse model in which APTX is deleted together with tyrosyl DNA phosphodiesterase (TDP1), an enzyme that repairs 3'-termini at a subset of single-strand breaks (SSBs), including those with 3'-topoisomerase-1 (Top1) peptide. Notably, the global rate of repair of oxidative and alkylation-induced SSBs was significantly slower in Tdp1(-/-)/Aptx(-/-) double knockout quiescent mouse astrocytes compared with Tdp1(-/-) or Aptx(-/-) single knockouts. In contrast, camptothecin-induced Top1-SSBs accumulated to similar levels in Tdp1(-/-) and Tdp1(-/-)/Aptx(-/-) double knockout astrocytes. Finally, we failed to identify a measurable defect in double-strand break repair in Tdp1(-/-), Aptx(-/-) or Tdp1(-/-)/Aptx(-/-) astrocytes. These data provide direct evidence for a requirement for aprataxin during chromosomal single-strand break repair in primary neural cells lacking Tdp1.
Figures

Similar articles
-
TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo.EMBO J. 2007 Nov 14;26(22):4720-31. doi: 10.1038/sj.emboj.7601869. Epub 2007 Oct 4. EMBO J. 2007. PMID: 17914460 Free PMC article.
-
TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1.Nucleic Acids Res. 2012 Sep 1;40(17):8371-80. doi: 10.1093/nar/gks622. Epub 2012 Jun 26. Nucleic Acids Res. 2012. PMID: 22740648 Free PMC article.
-
Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia 1.Mol Cell Biol. 2009 Mar;29(5):1354-62. doi: 10.1128/MCB.01471-08. Epub 2008 Dec 22. Mol Cell Biol. 2009. PMID: 19103743 Free PMC article.
-
Neurological disorders associated with DNA strand-break processing enzymes.Mech Ageing Dev. 2017 Jan;161(Pt A):130-140. doi: 10.1016/j.mad.2016.07.009. Epub 2016 Jul 25. Mech Ageing Dev. 2017. PMID: 27470939 Free PMC article. Review.
-
The role of TDP1 and APTX in mitochondrial DNA repair.Biochimie. 2014 May;100:121-4. doi: 10.1016/j.biochi.2013.10.011. Epub 2013 Oct 22. Biochimie. 2014. PMID: 24161509 Free PMC article. Review.
Cited by
-
Lack of aprataxin impairs mitochondrial functions via downregulation of the APE1/NRF1/NRF2 pathway.Hum Mol Genet. 2015 Aug 15;24(16):4516-29. doi: 10.1093/hmg/ddv183. Epub 2015 May 14. Hum Mol Genet. 2015. PMID: 25976310 Free PMC article.
-
The role of DNA base excision repair in brain homeostasis and disease.DNA Repair (Amst). 2015 Aug;32:172-179. doi: 10.1016/j.dnarep.2015.04.029. Epub 2015 May 1. DNA Repair (Amst). 2015. PMID: 26002197 Free PMC article. Review.
-
TORC2 inhibition triggers yeast chromosome fragmentation through misregulated Base Excision Repair of clustered oxidation events.Nat Commun. 2024 Nov 15;15(1):9908. doi: 10.1038/s41467-024-54142-z. Nat Commun. 2024. PMID: 39548071 Free PMC article.
-
Topoisomerase I inhibition in colorectal cancer: biomarkers and therapeutic targets.Br J Cancer. 2012 Jan 3;106(1):18-24. doi: 10.1038/bjc.2011.498. Epub 2011 Nov 22. Br J Cancer. 2012. PMID: 22108516 Free PMC article. Review.
-
Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2).DNA Repair (Amst). 2014 Jul;19:114-29. doi: 10.1016/j.dnarep.2014.03.020. Epub 2014 May 22. DNA Repair (Amst). 2014. PMID: 24856239 Free PMC article. Review.
References
-
- Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008;9:619–631. - PubMed
-
- Date H., Onodera O., Tanaka H., Iwabuchi K., Uekawa K., Igarashi S., Koike R., Hiroi T., Yuasa T., Awaya Y., Sakai T., Takahashi T., Nagatomo H., Sekijima Y., Kawachi I., Takiyama Y., Nishizawa M., Fukuhara N., Saito K., Sugano S., Tsuji S. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001;29:184–188. - PubMed
-
- Moreira M.C., Barbot C., Tachi N., Kozuka N., Uchida E., Gibson T., Mendonca P., Costa M., Barros J., Yanagisawa T., Watanabe M., Ikeda Y., Aoki M., Nagata T., Coutinho P., Sequeiros J., Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001;29:189–193. - PubMed
-
- Lavin M.F., Gueven N., Grattan-Smith P. Defective responses to DNA single- and double-strand breaks in spinocerebellar ataxia. DNA Repair (Amst.) 2008;7:1061–1076. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
