

Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase
- PMID: 19304573
- PMCID: PMC2811263
- DOI: 10.1161/ATVBAHA.109.186577
Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase
Abstract
Objective: GPIHBP1 is an endothelial cell protein that binds lipoprotein lipase (LPL) and chylomicrons. Because GPIHBP1 deficiency causes chylomicronemia in mice, we sought to determine whether some cases of chylomicronemia in humans could be attributable to defective GPIHBP1 proteins.
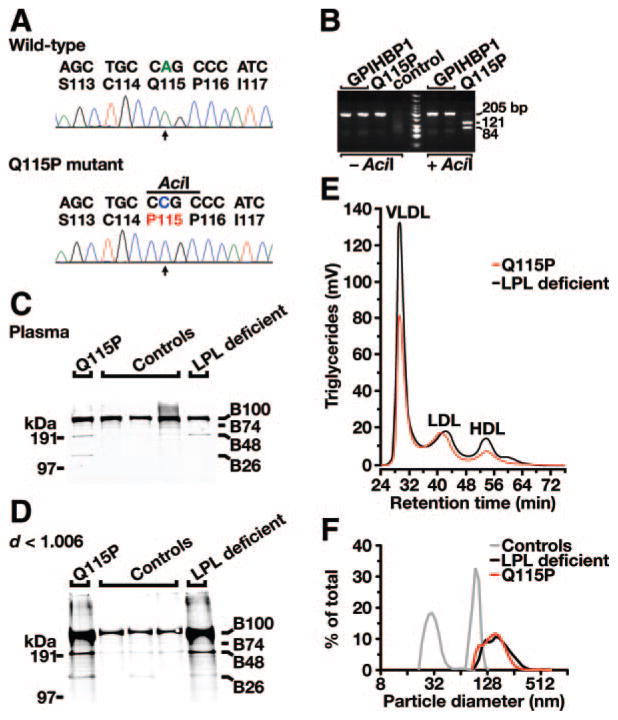
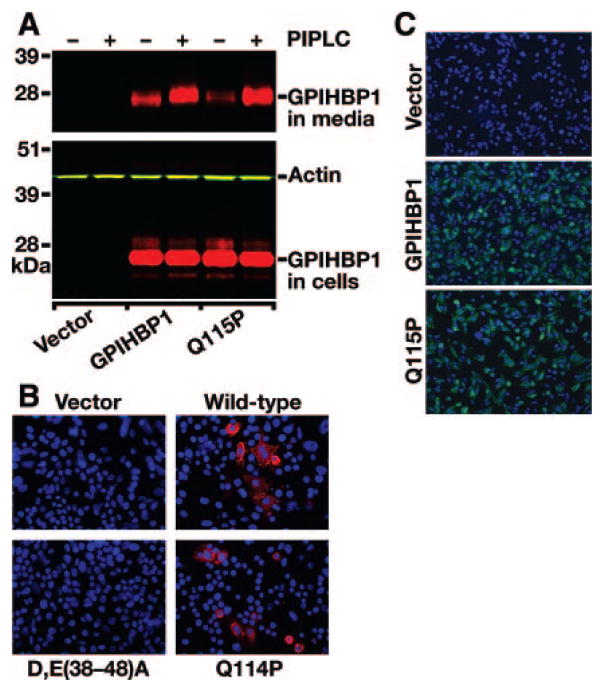
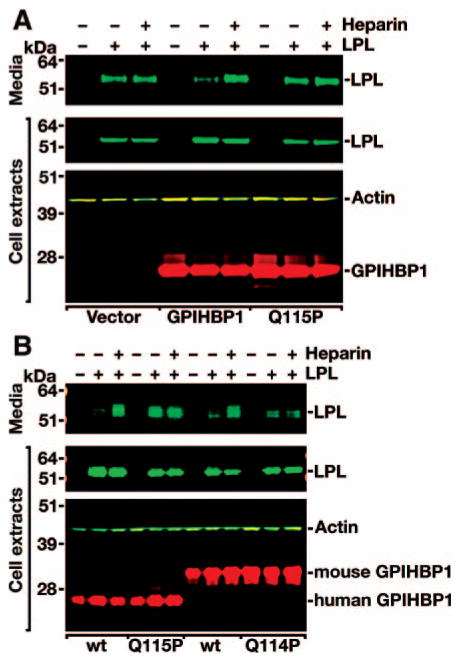
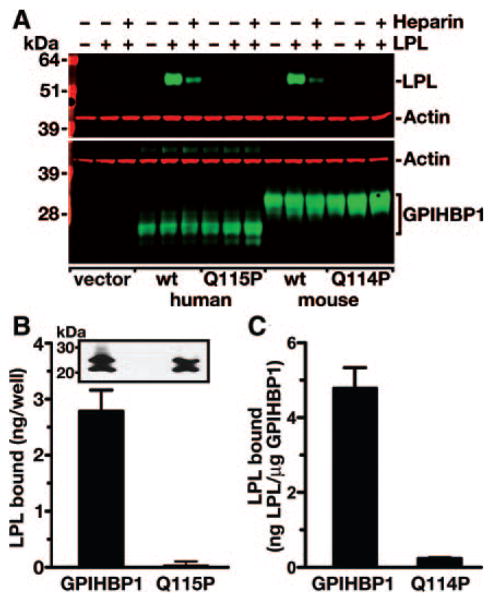
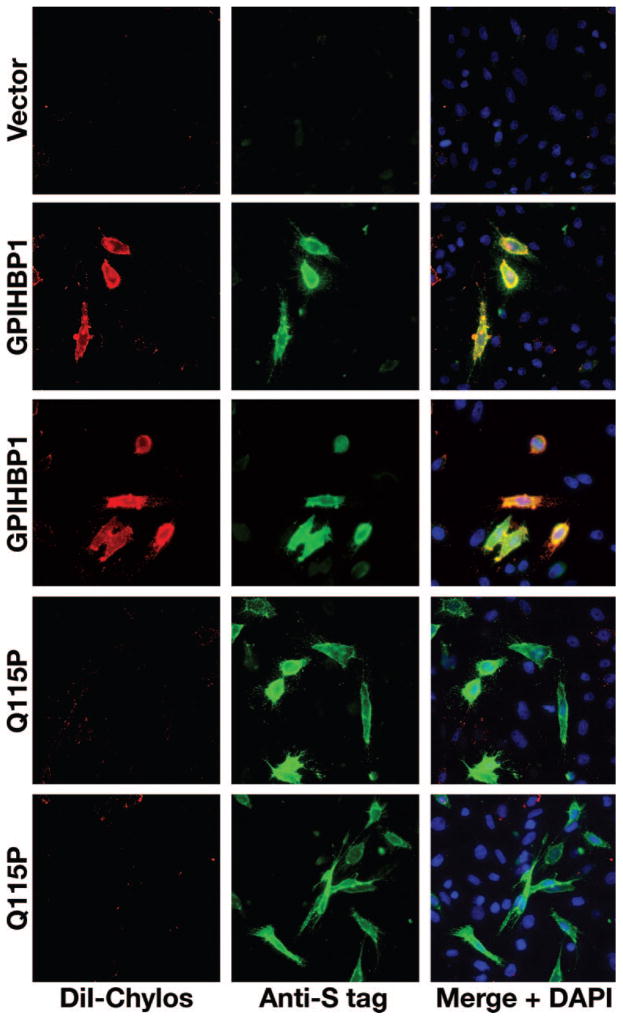
Methods and results: Patients with severe hypertriglyceridemia (n=60, with plasma triglycerides above the 95th percentile for age and gender) were screened for mutations in GPIHBP1. A homozygous GPIHBP1 mutation (c.344A>C) that changed a highly conserved glutamine at residue 115 to a proline (p.Q115P) was identified in a 33-year-old male with lifelong chylomicronemia. The patient had failure-to-thrive as a child but had no history of pancreatitis. He had no mutations in LPL, APOA5, or APOC2. The Q115P substitution did not affect the ability of GPIHBP1 to reach the cell surface. However, unlike wild-type GPIHBP1, GPIHBP1-Q115P lacked the ability to bind LPL or chylomicrons (d < 1.006 g/mL lipoproteins from Gpihbp1(-/-) mice). Mouse GPIHBP1 with the corresponding mutation (Q114P) also could not bind LPL.
Conclusions: A homozygous missense mutation in GPIHBP1 (Q115P) was identified in a patient with chylomicronemia. The mutation eliminated the ability of GPIHBP1 to bind LPL and chylomicrons, strongly suggesting that it caused the patient's chylomicronemia.
Figures
Comment in
-
Some things just have to be done in vivo: GPIHBP1, caloric delivery, and the generation of remnant lipoproteins.Arterioscler Thromb Vasc Biol. 2009 Jun;29(6):792-5. doi: 10.1161/ATVBAHA.109.187823. Arterioscler Thromb Vasc Biol. 2009. PMID: 19458350
References
-
- Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001.
-
- Beigneux AP, Davies B, Gin P, Weinstein MM, Farber E, Qiao X, Peale P, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. - PMC - PubMed
-
- Havel RJ, Kane JP. Introduction: Structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
