

Pervasive, genome-wide positive selection leading to functional divergence in the bacterial genus Campylobacter
- PMID: 19304960
- PMCID: PMC2704436
- DOI: 10.1101/gr.089250.108
Pervasive, genome-wide positive selection leading to functional divergence in the bacterial genus Campylobacter
Abstract
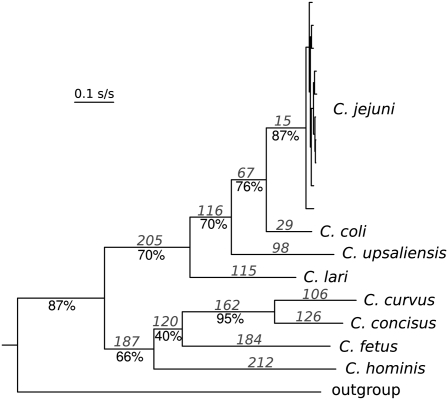
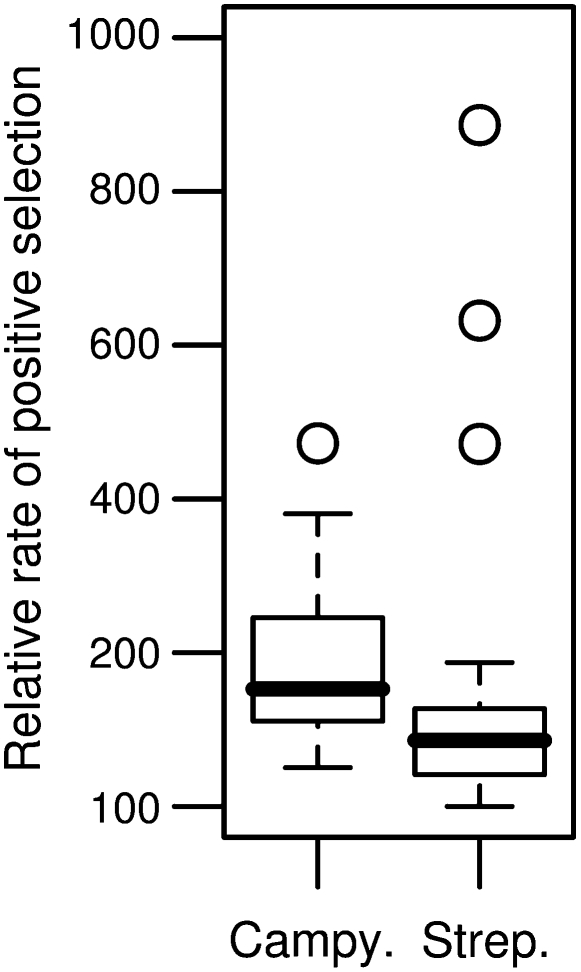
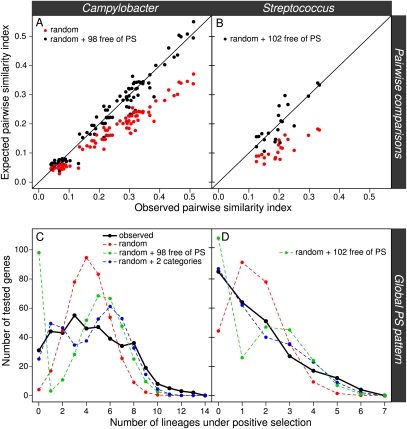
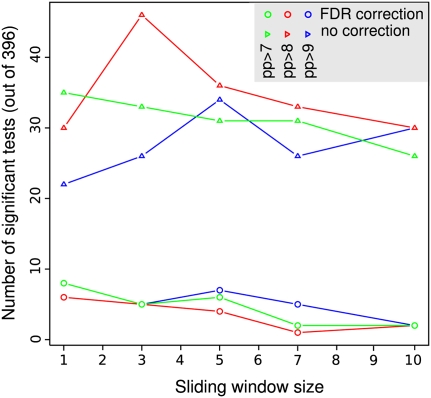
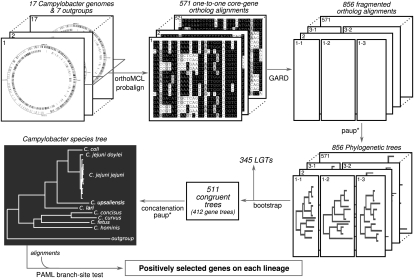
An open question in bacterial genomics is the role that adaptive evolution of the core genome plays in diversification and adaptation of bacterial species, and how this might differ between groups of bacteria occupying different environmental circumstances. The genus Campylobacter encompasses several important human and animal enteric pathogens, with genome sequence data available for eight species. We estimate the Campylobacter core genome at 647 genes, with 92.5% of the nonrecombinant core genome loci under positive selection in at least one lineage and the same gene frequently under positive selection in multiple lineages. Tests are provided that reject recombination, saturation, and variation in codon usage bias as factors contributing to this high level of selection. We suggest this genome-wide adaptive evolution may result from a Red Queen macroevolutionary dynamic, in which species are involved in competition for resources within the mammalian and/or vertebrate gastrointestinal tract. Much reduced levels of positive selection evident in Streptococcus, as reported by the authors in an earlier work, may be a consequence of these taxa inhabiting less species-rich habitats, and more unique niches. Despite many common loci under positive selection in multiple Campylobacter lineages, we found no evidence for molecular adaptive convergence at the level of the same or adjacent codons, or even protein domains. Taken collectively, these results describe the diversification of a bacterial genus that involves pervasive natural selection pressure across virtually the entire genome, with this adaptation occurring in different ways in different lineages, despite the species tendency toward a common gastrointestinal habitat.
Figures

Similar articles
-
Genome-wide analyses reveal lineage specific contributions of positive selection and recombination to the evolution of Listeria monocytogenes.BMC Evol Biol. 2008 Aug 12;8:233. doi: 10.1186/1471-2148-8-233. BMC Evol Biol. 2008. PMID: 18700032 Free PMC article.
-
Natural selection and recombination at host-interacting lipoprotein loci drive genome diversification of Lyme disease and related bacteria.mBio. 2024 Sep 11;15(9):e0174924. doi: 10.1128/mbio.01749-24. Epub 2024 Aug 15. mBio. 2024. PMID: 39145656 Free PMC article.
-
Genomic evidence for the emergence and evolution of pathogenicity and niche preferences in the genus Campylobacter.Genome Biol Evol. 2014 Sep 4;6(9):2392-405. doi: 10.1093/gbe/evu195. Genome Biol Evol. 2014. PMID: 25193310 Free PMC article.
-
The genomics of organismal diversification illuminated by adaptive radiations.Trends Genet. 2015 Sep;31(9):491-9. doi: 10.1016/j.tig.2015.07.002. Epub 2015 Aug 7. Trends Genet. 2015. PMID: 26259669 Review.
-
Using genome scans of DNA polymorphism to infer adaptive population divergence.Mol Ecol. 2005 Mar;14(3):671-88. doi: 10.1111/j.1365-294X.2005.02437.x. Mol Ecol. 2005. PMID: 15723660 Review.
Cited by
-
Comparative genomics Lactobacillus reuteri from sourdough reveals adaptation of an intestinal symbiont to food fermentations.Sci Rep. 2015 Dec 11;5:18234. doi: 10.1038/srep18234. Sci Rep. 2015. PMID: 26658825 Free PMC article.
-
Introgression in the genus Campylobacter: generation and spread of mosaic alleles.Microbiology (Reading). 2011 Apr;157(Pt 4):1066-1074. doi: 10.1099/mic.0.045153-0. Epub 2011 Jan 6. Microbiology (Reading). 2011. PMID: 21212120 Free PMC article.
-
Pan-Genome Analysis of Campylobacter: Insights on the Genomic Diversity and Virulence Profile.Microbiol Spectr. 2022 Oct 26;10(5):e0102922. doi: 10.1128/spectrum.01029-22. Epub 2022 Sep 7. Microbiol Spectr. 2022. PMID: 36069574 Free PMC article.
-
Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage.PLoS One. 2011;6(11):e27121. doi: 10.1371/journal.pone.0027121. Epub 2011 Nov 11. PLoS One. 2011. PMID: 22096527 Free PMC article.
-
Genome-Wide Analyses Reveal Genes Subject to Positive Selection in Pasteurella multocida.Front Microbiol. 2017 May 30;8:961. doi: 10.3389/fmicb.2017.00961. eCollection 2017. Front Microbiol. 2017. PMID: 28611758 Free PMC article.
References
-
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statist. 2001;29(Suppl. 4):1165–1188.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources