

Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1
- PMID: 19305497
- PMCID: PMC2654723
- DOI: 10.1371/journal.pone.0004954
Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1
Abstract
Background: Palmitate is a potent inducer of endoplasmic reticulum (ER) stress in beta-cells. In type 2 diabetes, glucose amplifies fatty-acid toxicity for pancreatic beta-cells, leading to beta-cell dysfunction and death. Why glucose exacerbates beta-cell lipotoxicity is largely unknown. Glucose stimulates mTORC1, an important nutrient sensor involved in the regulation of cellular stress. Our study tested the hypothesis that glucose augments lipotoxicity by stimulating mTORC1 leading to increased beta-cell ER stress.
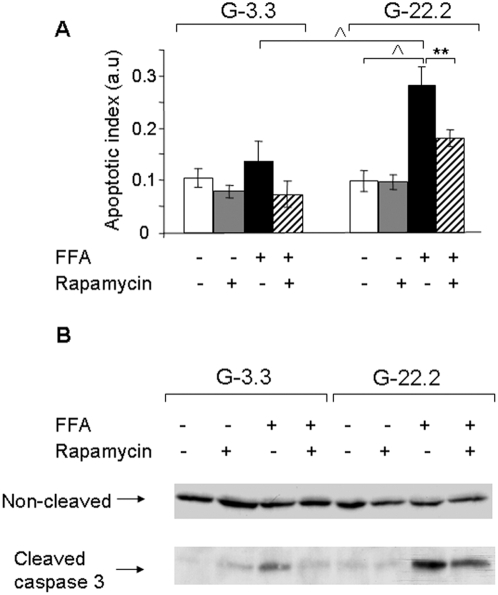
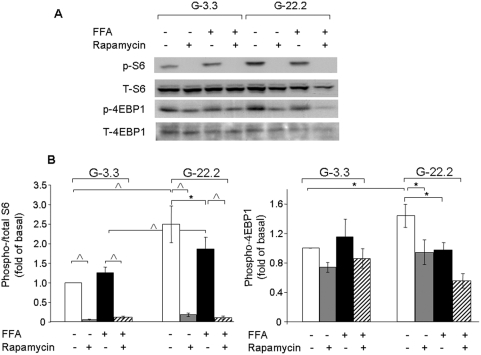
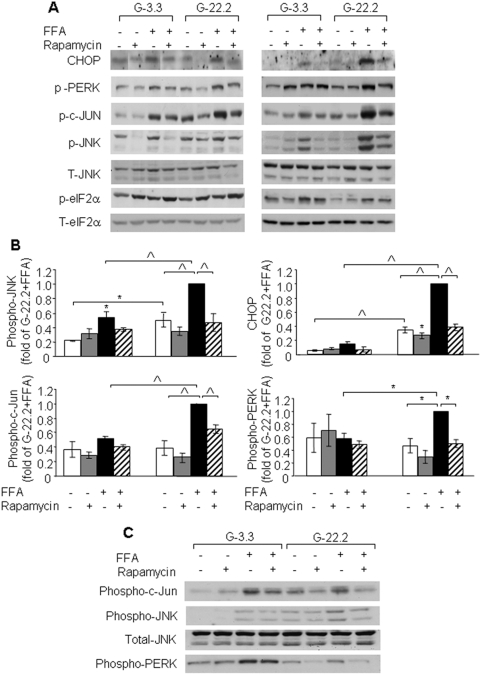
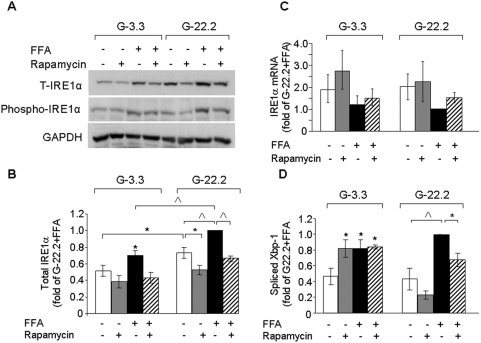
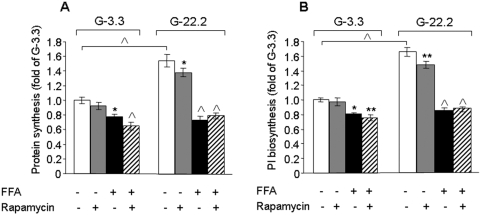
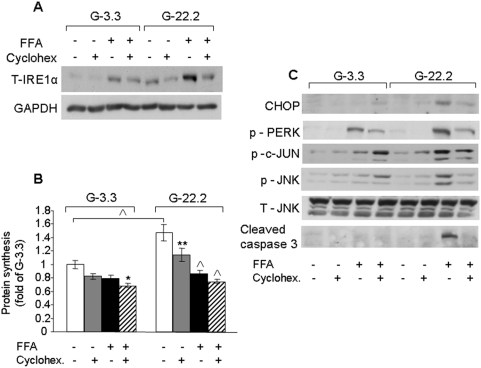
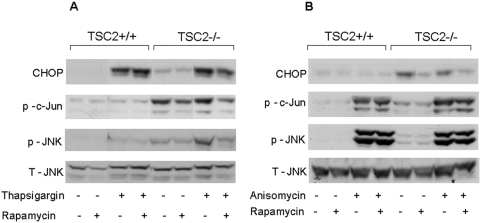
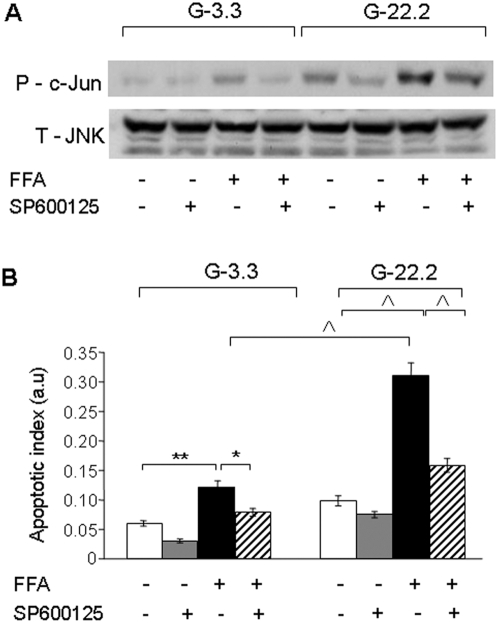
Principal findings: We found that glucose amplifies palmitate-induced ER stress by increasing IRE1alpha protein levels and activating the JNK pathway, leading to increased beta-cell apoptosis. Moreover, glucose increased mTORC1 activity and its inhibition by rapamycin decreased beta-cell apoptosis under conditions of glucolipotoxicity. Inhibition of mTORC1 by rapamycin did not affect proinsulin and total protein synthesis in beta-cells incubated at high glucose with palmitate. However, it decreased IRE1alpha expression and signaling and inhibited JNK pathway activation. In TSC2-deficient mouse embryonic fibroblasts, in which mTORC1 is constitutively active, mTORC1 regulated the stimulation of JNK by ER stressors, but not in response to anisomycin, which activates JNK independent of ER stress. Finally, we found that JNK inhibition decreased beta-cell apoptosis under conditions of glucolipotoxicity.
Conclusions/significance: Collectively, our findings suggest that mTORC1 mediates glucose amplification of lipotoxicity, acting through activation of ER stress and JNK. Thus, mTORC1 is an important transducer of ER stress in beta-cell glucolipotoxicity. Moreover, in stressed beta-cells mTORC1 inhibition decreases IRE1alpha protein expression and JNK activity without affecting ER protein load, suggesting that mTORC1 regulates the beta-cell stress response to glucose and fatty acids by modulating the synthesis and activity of specific proteins involved in the execution of the ER stress response. This novel paradigm may have important implications for understanding beta-cell failure in type 2 diabetes.
Conflict of interest statement
Figures
References
-
- Goh TT, Mason TM, Gupta N, So A, Lam TK, et al. Lipid-induced beta-cell dysfunction in vivo in models of progressive beta-cell failure. Am J Physiol Endocrinol Metab. 2007;292:E549–E560. - PubMed
-
- Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, et al. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes. 2001;59:69–76. - PubMed
-
- Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52:726–733. - PubMed
-
- Eitel K, Staiger H, Brendel MD, Brandhorst D, Bretzel RG, et al. Different role of saturated and unsaturated fatty acids in beta-cell apoptosis. Biochem Biophys Res Commun. 2002;299:853–856. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
