

Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing
- PMID: 19307598
- PMCID: PMC2699156
- DOI: 10.1083/jcb.200809028
Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing
Abstract
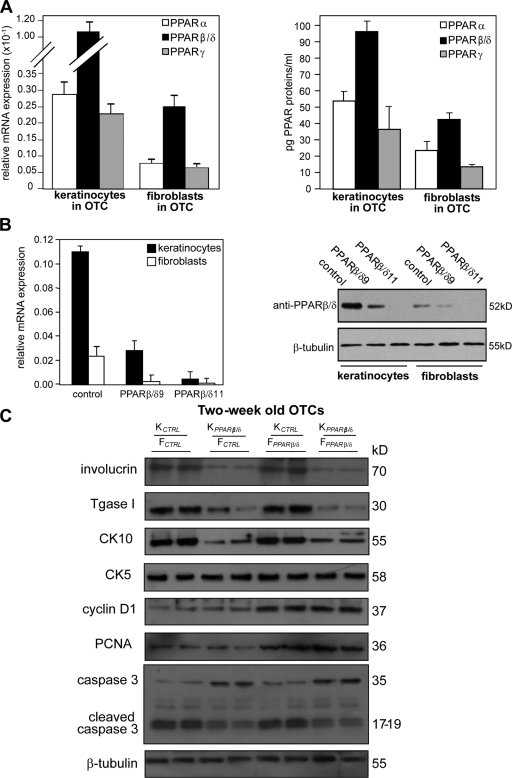
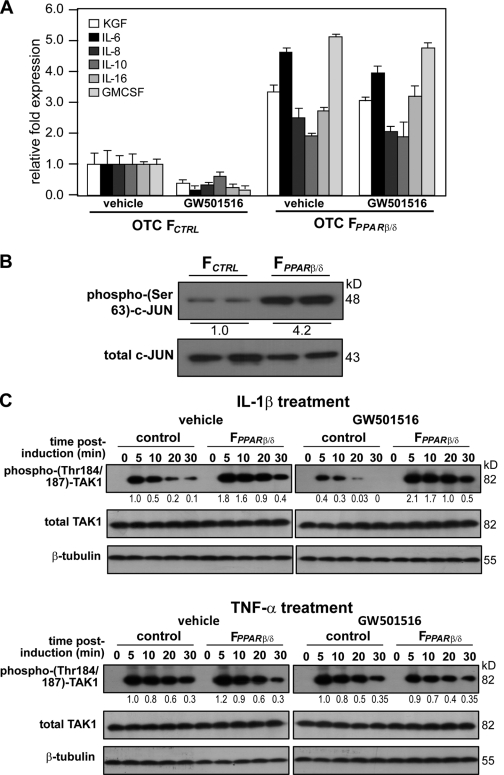
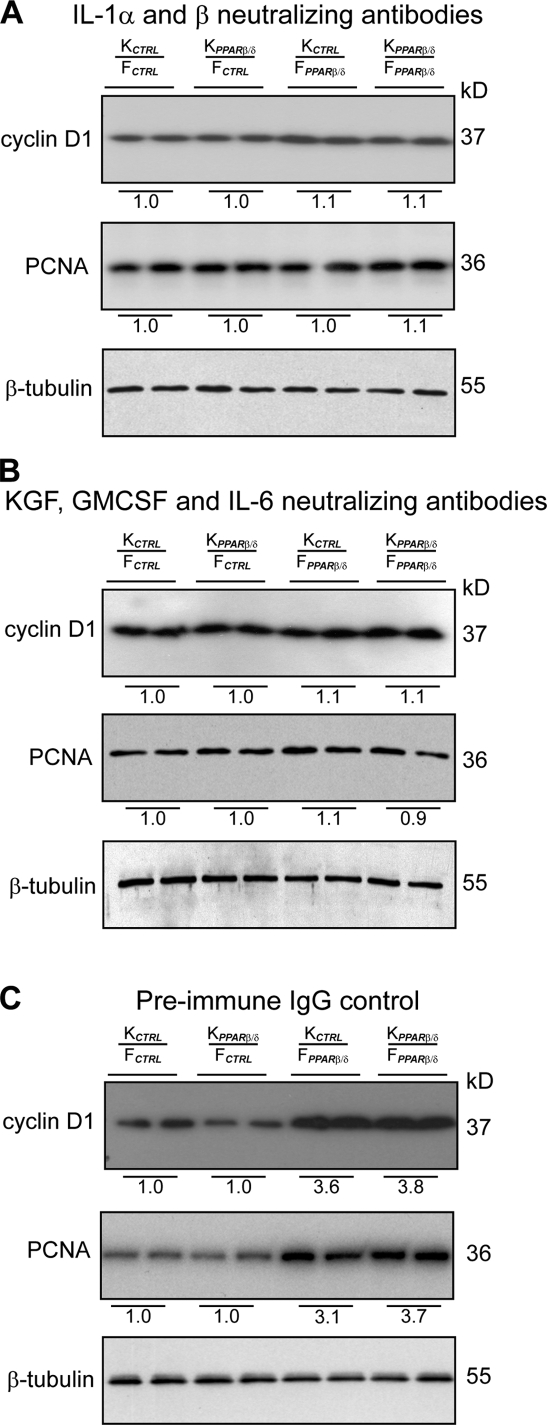
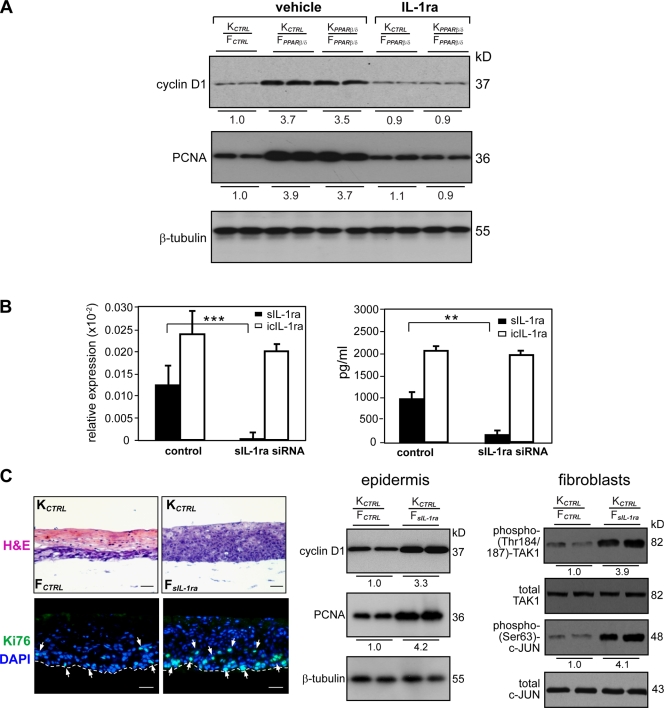
Skin morphogenesis, maintenance, and healing after wounding require complex epithelial-mesenchymal interactions. In this study, we show that for skin homeostasis, interleukin-1 (IL-1) produced by keratinocytes activates peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) expression in underlying fibroblasts, which in turn inhibits the mitotic activity of keratinocytes via inhibition of the IL-1 signaling pathway. In fact, PPARbeta/delta stimulates production of the secreted IL-1 receptor antagonist, which leads to an autocrine decrease in IL-1 signaling pathways and consequently decreases production of secreted mitogenic factors by the fibroblasts. This fibroblast PPARbeta/delta regulation of the IL-1 signaling is required for proper wound healing and can regulate tumor as well as normal human keratinocyte cell proliferation. Together, these findings provide evidence for a novel homeostatic control of keratinocyte proliferation and differentiation mediated via PPARbeta/delta regulation in dermal fibroblasts of IL-1 signaling. Given the ubiquitous expression of PPARbeta/delta, other epithelial-mesenchymal interactions may also be regulated in a similar manner.
Figures

Similar articles
-
Time (and PPAR beta/delta) heals all wounds.J Cell Biol. 2009 Mar 23;184(6):767. doi: 10.1083/jcb.1846if. J Cell Biol. 2009. PMID: 19307594 Free PMC article.
-
Epithelium-mesenchyme interactions control the activity of peroxisome proliferator-activated receptor beta/delta during hair follicle development.Mol Cell Biol. 2005 Mar;25(5):1696-712. doi: 10.1128/MCB.25.5.1696-1712.2005. Mol Cell Biol. 2005. PMID: 15713628 Free PMC article.
-
Rosiglitazone induces interleukin-1 receptor antagonist in interleukin-1beta-stimulated rat synovial fibroblasts via a peroxisome proliferator-activated receptor beta/delta-dependent mechanism.Arthritis Rheum. 2005 Mar;52(3):759-69. doi: 10.1002/art.20868. Arthritis Rheum. 2005. PMID: 15751073
-
Nuclear receptor peroxisome proliferator activated receptor (PPAR) β/δ in skin wound healing and cancer.Eur J Dermatol. 2015 Apr;25 Suppl 1:4-11. doi: 10.1684/ejd.2014.2505. Eur J Dermatol. 2015. PMID: 26287030 Review.
-
Regulatory mechanisms mediated by peroxisome proliferator-activated receptor-β/δ in skin cancer.Mol Carcinog. 2019 Sep;58(9):1612-1622. doi: 10.1002/mc.23033. Epub 2019 May 6. Mol Carcinog. 2019. PMID: 31062422 Free PMC article. Review.
Cited by
-
Hepatoprotective effect of resveratrol against ethanol-induced oxidative stress through induction of superoxide dismutase in vivo and in vitro.Exp Ther Med. 2016 Apr;11(4):1231-1238. doi: 10.3892/etm.2016.3077. Epub 2016 Feb 16. Exp Ther Med. 2016. PMID: 27073428 Free PMC article.
-
PPARβ/δ: Linking Metabolism to Regeneration.Int J Mol Sci. 2018 Jul 10;19(7):2013. doi: 10.3390/ijms19072013. Int J Mol Sci. 2018. PMID: 29996502 Free PMC article. Review.
-
PPARδ agonist GW501516 inhibits PDGF-stimulated pulmonary arterial smooth muscle cell function related to pathological vascular remodeling.Biomed Res Int. 2013;2013:903947. doi: 10.1155/2013/903947. Epub 2013 Mar 27. Biomed Res Int. 2013. PMID: 23607100 Free PMC article.
-
Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration.Am J Pathol. 2010 Dec;177(6):2791-803. doi: 10.2353/ajpath.2010.100129. Epub 2010 Oct 15. Am J Pathol. 2010. PMID: 20952587 Free PMC article.
-
Cytokines and Regulating Epithelial Cell Division.Curr Drug Targets. 2024;25(3):190-200. doi: 10.2174/0113894501279979240101051345. Curr Drug Targets. 2024. PMID: 38213162 Review.
References
-
- Arend W.P., Malyak M., Guthridge C.J., Gabay C. 1998. Interleukin-1 receptor antagonist: role in biology.Annu. Rev. Immunol. 16:27–55 - PubMed
-
- Banda N.K., Guthridge C., Sheppard D., Cairns K.S., Muggli M., Bech-Otschir D., Dubiel W., Arend W.P. 2005. Intracellular IL-1 receptor antagonist type 1 inhibits IL-1-induced cytokine production in keratinocytes through binding to the third component of the COP9 signalosome.J. Immunol. 174:3608–3616 - PubMed
-
- Beanes S.R., Dang C., Soo C., Ting K. 2003. Skin repair and scar formation: the central role of TGF-beta.Expert Rev. Mol. Med. 5:1–22 - PubMed
-
- Burdick A.D., Kim D.J., Peraza M.A., Gonzalez F.J., Peters J.M. 2006. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation.Cell. Signal. 18:9–20 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
