

Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability
- PMID: 19307602
- PMCID: PMC2699149
- DOI: 10.1083/jcb.200805063
Akt regulates L-type Ca2+ channel activity by modulating Cavalpha1 protein stability
Erratum in
- J Cell Biol. 2013 Mar 18;200(6):851
Abstract
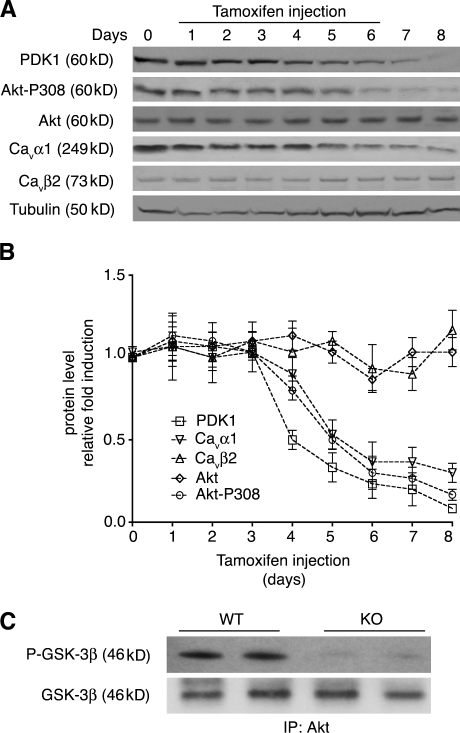
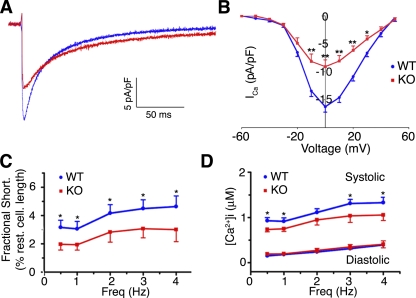
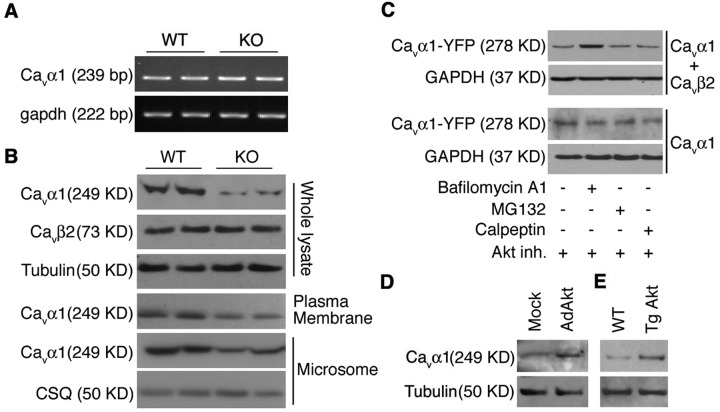
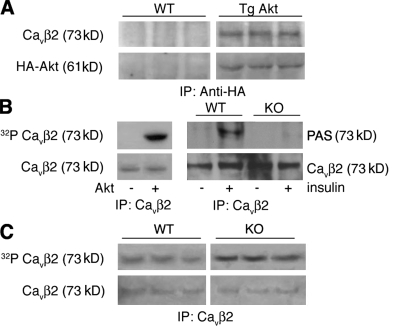
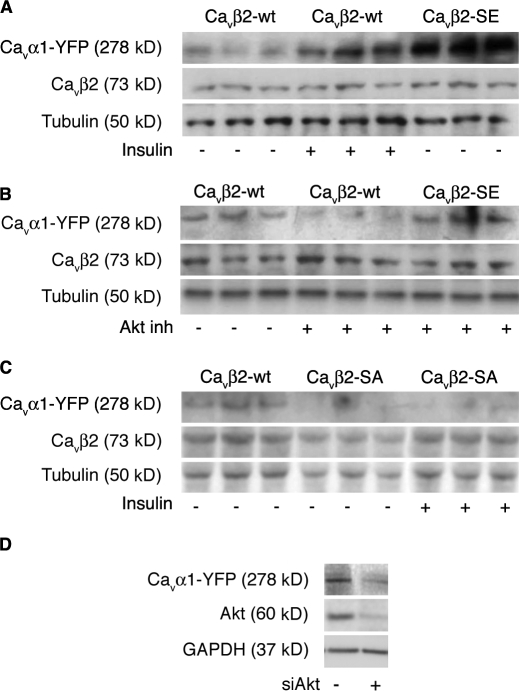
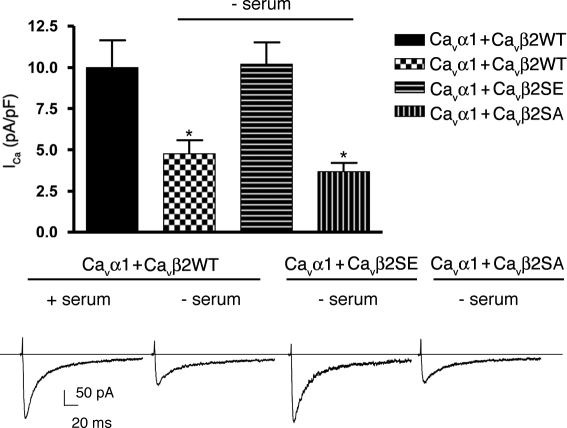
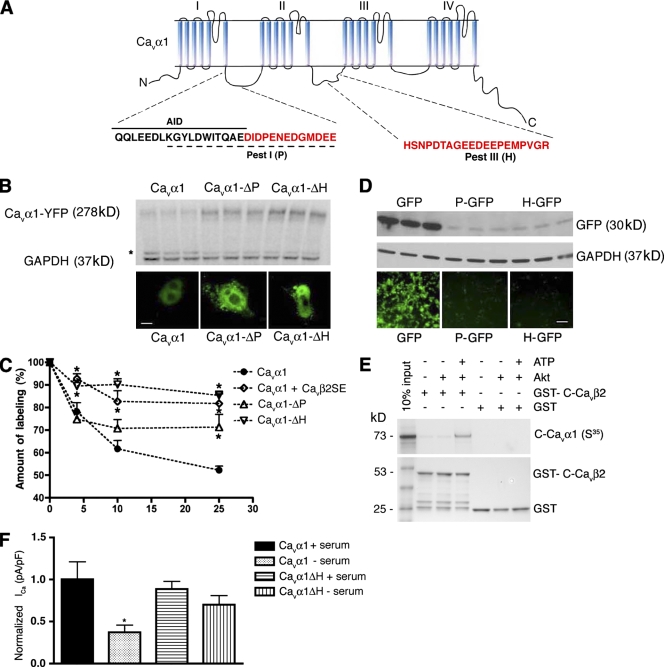
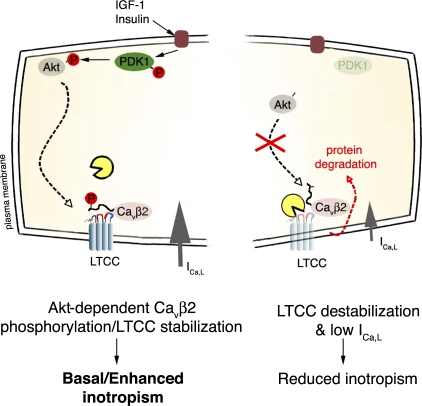
The insulin IGF-1-PI3K-Akt signaling pathway has been suggested to improve cardiac inotropism and increase Ca(2+) handling through the effects of the protein kinase Akt. However, the underlying molecular mechanisms remain largely unknown. In this study, we provide evidence for an unanticipated regulatory function of Akt controlling L-type Ca(2+) channel (LTCC) protein density. The pore-forming channel subunit Ca(v)alpha1 contains highly conserved PEST sequences (signals for rapid protein degradation), and in-frame deletion of these PEST sequences results in increased Ca(v)alpha1 protein levels. Our findings show that Akt-dependent phosphorylation of Ca(v)beta2, the LTCC chaperone for Ca(v)alpha1, antagonizes Ca(v)alpha1 protein degradation by preventing Ca(v)alpha1 PEST sequence recognition, leading to increased LTCC density and the consequent modulation of Ca(2+) channel function. This novel mechanism by which Akt modulates LTCC stability could profoundly influence cardiac myocyte Ca(2+) entry, Ca(2+) handling, and contractility.
Figures
References
-
- Aimond F., Kwak S.P., Rhodes K.J., Nerbonne J.M. 2005. Accessory Kvbeta1 subunits differentially modulate the functional expression of voltage-gated K+ channels in mouse ventricular myocytes. Circ. Res. 96:451–458 - PubMed
-
- Belles B., Hescheler J., Trautwein W., Blomgren K., Karlsson J.O. 1988. A possible physiological role of the Ca-dependent protease calpain and its inhibitor calpastatin on the Ca current in guinea pig myocytes. Pflugers Arch. 412:554–556 - PubMed
-
- Bers D.M. 2002. Cardiac excitation-contraction coupling. Nature. 415:198–205 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
