

Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway
- PMID: 19307610
- PMCID: PMC2784410
- DOI: 10.1165/rcmb.2008-0157OC
Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway
Abstract
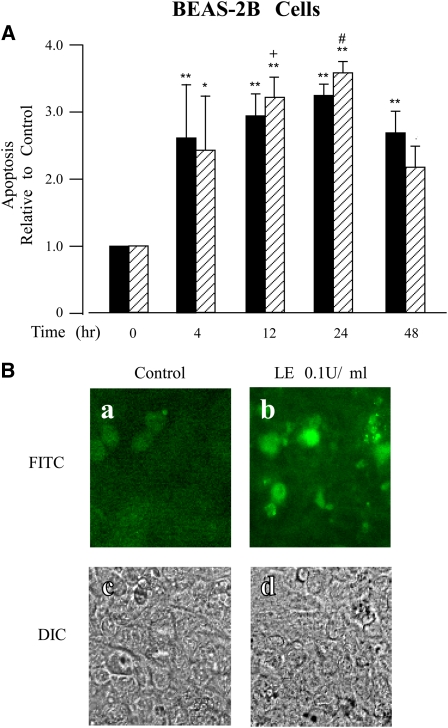
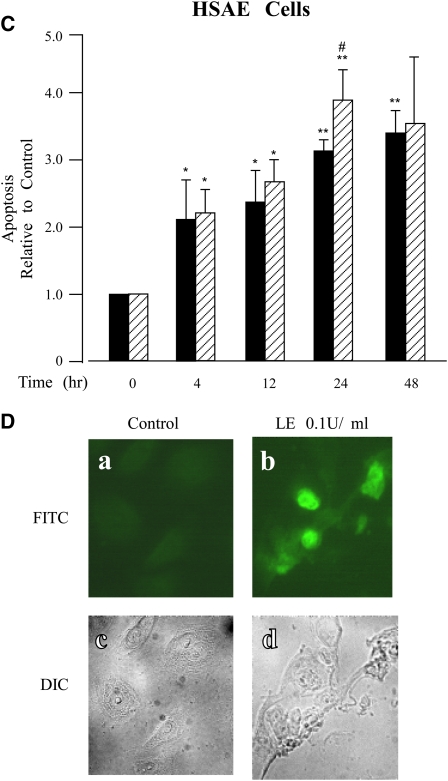
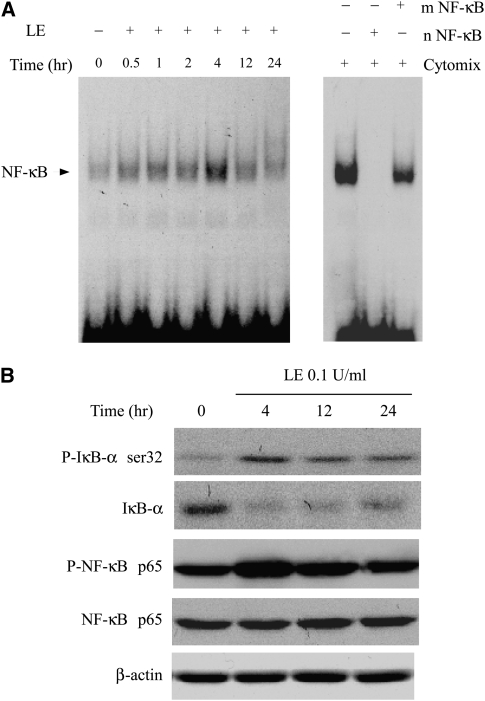
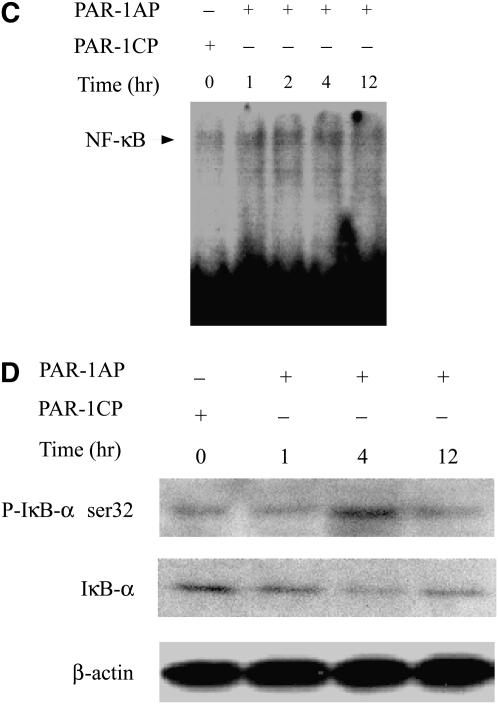
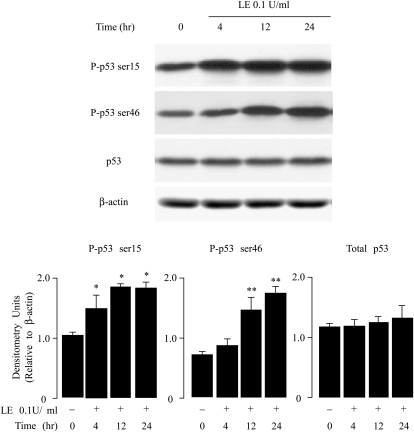
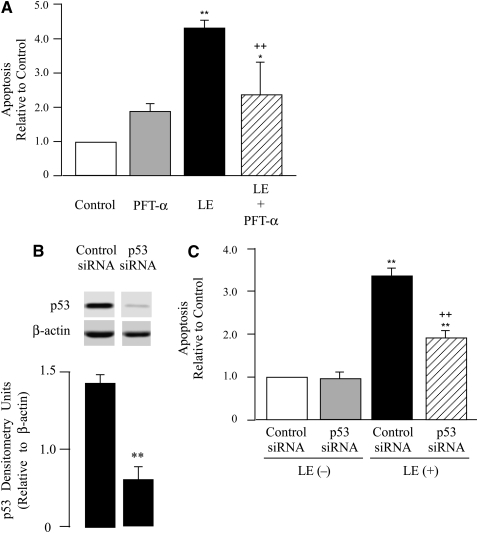
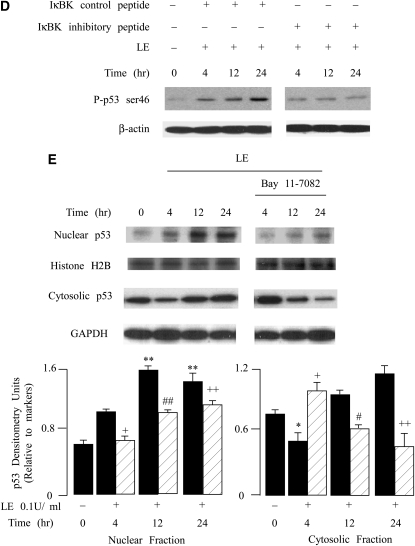
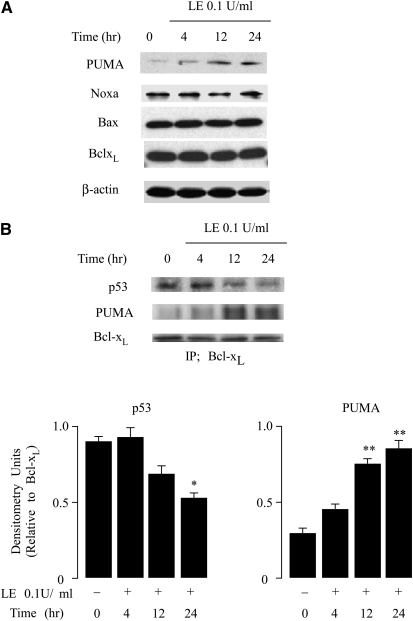
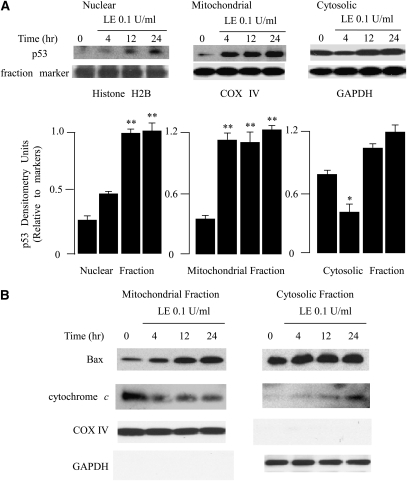
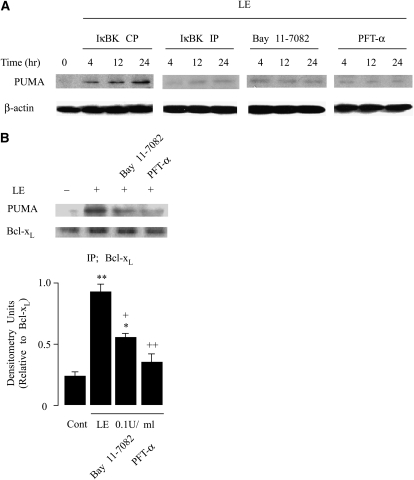
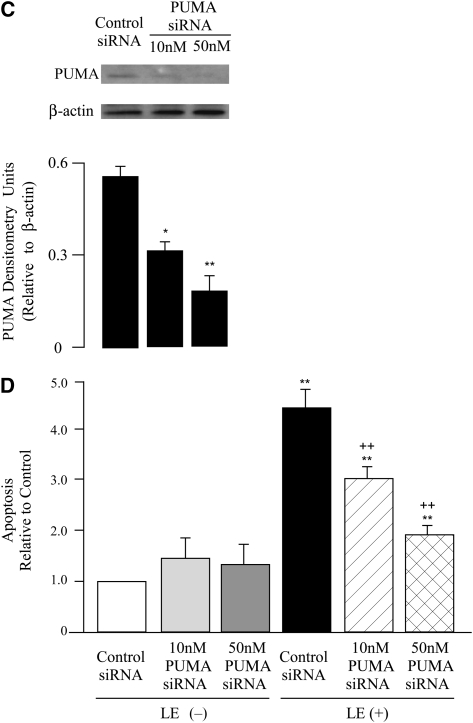
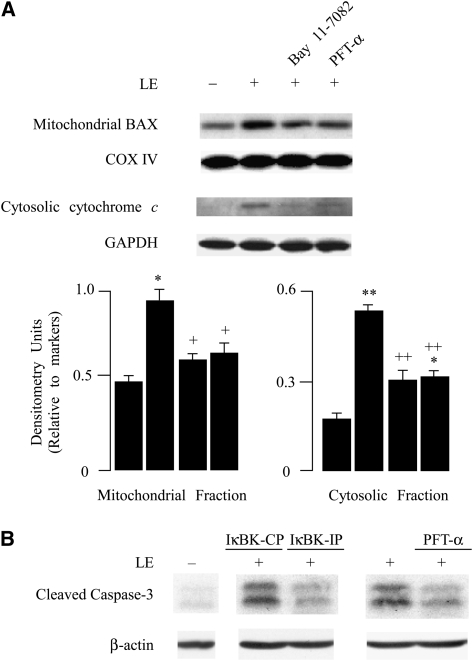
Leukocyte elastase induces apoptosis of lung epithelial cells via alterations in mitochondrial permeability, but the signaling pathways regulating this response remain uncertain. Here we investigated the involvement of proteinase-activated receptor-1 (PAR-1), the transcription factor NF-kappaB, and the protooncogene p53 in this pathway. Elastase-induced apoptosis of lung epithelial cells correlated temporally with activation of NF-kappaB, phosphorylation, and nuclear translocation of p53, increased p53 up-regulated modulator of apoptosis (PUMA) expression, and mitochondrial translocation of Bax resulting in enhanced permeability. Elastase-induced apoptosis was also prevented by pharmacologic inhibitors of NF-kappaB and p53 and by short interfering RNA knockdown of PAR-1, p53, or PUMA. These inhibitors prevented elastase-induced PUMA expression, mitochondrial translocation of Bax, increased mitochondrial permeability, and attenuated apoptosis. NF-kappaB inhibitors also reduced p53 phosphorylation. We conclude that elastase-induced apoptosis of lung epithelial cells is mediated by a PAR-1-triggered pathway involving activation of NF-kappaB and p53, and a PUMA- and Bax-dependent increase in mitochondrial permeability leading to activation of distal caspases. Further, p53 contributes to elastase-induced apoptosis by both transcriptional and post-transcriptional mechanisms.
Figures
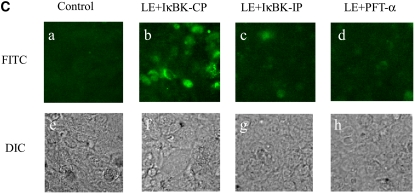
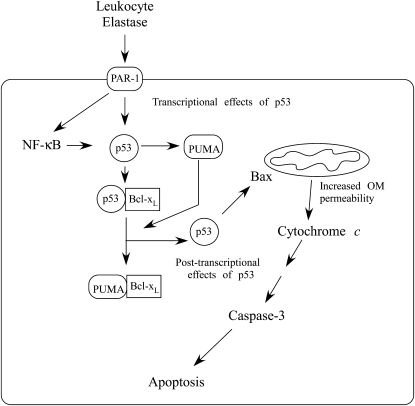
Similar articles
-
Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells.Am J Respir Cell Mol Biol. 2005 Sep;33(3):231-47. doi: 10.1165/rcmb.2005-0109OC. Epub 2005 May 12. Am J Respir Cell Mol Biol. 2005. PMID: 15891109 Free PMC article.
-
Inhibiting aberrant p53-PUMA feedback loop activation attenuates ischaemia reperfusion-induced neuroapoptosis and neuroinflammation in rats by downregulating caspase 3 and the NF-κB cytokine pathway.J Neuroinflammation. 2018 Sep 1;15(1):250. doi: 10.1186/s12974-018-1271-9. J Neuroinflammation. 2018. PMID: 30172256 Free PMC article.
-
Role of p53, PUMA, and Bax in wogonin-induced apoptosis in human cancer cells.Biochem Pharmacol. 2008 May 15;75(10):2020-33. doi: 10.1016/j.bcp.2008.02.023. Epub 2008 Feb 29. Biochem Pharmacol. 2008. PMID: 18377871 Free PMC article.
-
The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases.Apoptosis. 2021 Jun;26(5-6):235-247. doi: 10.1007/s10495-021-01667-z. Epub 2021 Mar 30. Apoptosis. 2021. PMID: 33783663 Free PMC article. Review.
-
Nuclear factor-κB, p53, and mitochondria: regulation of cellular metabolism and the Warburg effect.Trends Biochem Sci. 2012 Aug;37(8):317-24. doi: 10.1016/j.tibs.2012.04.002. Epub 2012 May 23. Trends Biochem Sci. 2012. PMID: 22626470 Review.
Cited by
-
Thrombin induces macrophage migration inhibitory factor release and upregulation in urothelium: a possible contribution to bladder inflammation.PLoS One. 2010 Dec 31;5(12):e15904. doi: 10.1371/journal.pone.0015904. PLoS One. 2010. PMID: 21209875 Free PMC article.
-
Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2).J Biol Chem. 2011 Jul 15;286(28):24638-48. doi: 10.1074/jbc.M110.201988. Epub 2011 May 16. J Biol Chem. 2011. PMID: 21576245 Free PMC article.
-
Targeting neutrophil extracellular traps in severe acute pancreatitis treatment.Therap Adv Gastroenterol. 2020 Nov 24;13:1756284820974913. doi: 10.1177/1756284820974913. eCollection 2020. Therap Adv Gastroenterol. 2020. PMID: 33281940 Free PMC article. Review.
-
Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation.Sci Rep. 2016 May 31;6:24708. doi: 10.1038/srep24708. Sci Rep. 2016. PMID: 27241590 Free PMC article.
-
Glycosaminoglycans as Multifunctional Anti-Elastase and Anti-Inflammatory Drugs in Cystic Fibrosis Lung Disease.Front Pharmacol. 2020 Jul 8;11:1011. doi: 10.3389/fphar.2020.01011. eCollection 2020. Front Pharmacol. 2020. PMID: 32733248 Free PMC article. Review.
References
-
- Savill J. Apoptosis and renal injury. Curr Opin Nephrol Hypertens 1995;4:263–269. - PubMed
-
- Sabbah HN, Sharov VG. Apoptosis in heart failure. Prog Cardiovasc Dis 1998;40:549–562. - PubMed
-
- Ginzberg HH, Shannon PT, Suzuki T, Hong O, Vachon E, Moraes T, Abreu MT, Cherepanov V, Wang X, Chow CW, et al. Leukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and Akt. Am J Physiol Gastrointest Liver Physiol 2004;287:G286–G298. - PubMed
-
- Chin AC, Parkos CA. Pathobiology of neutrophil transepithelial migration: implications in mediating epithelial injury. Annu Rev Pathol 2007;2:111–143. - PubMed
-
- Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 2002;451:1–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
