

Functional aspects of protein flexibility
- PMID: 19308324
- PMCID: PMC11115794
- DOI: 10.1007/s00018-009-0014-6
Functional aspects of protein flexibility
Abstract
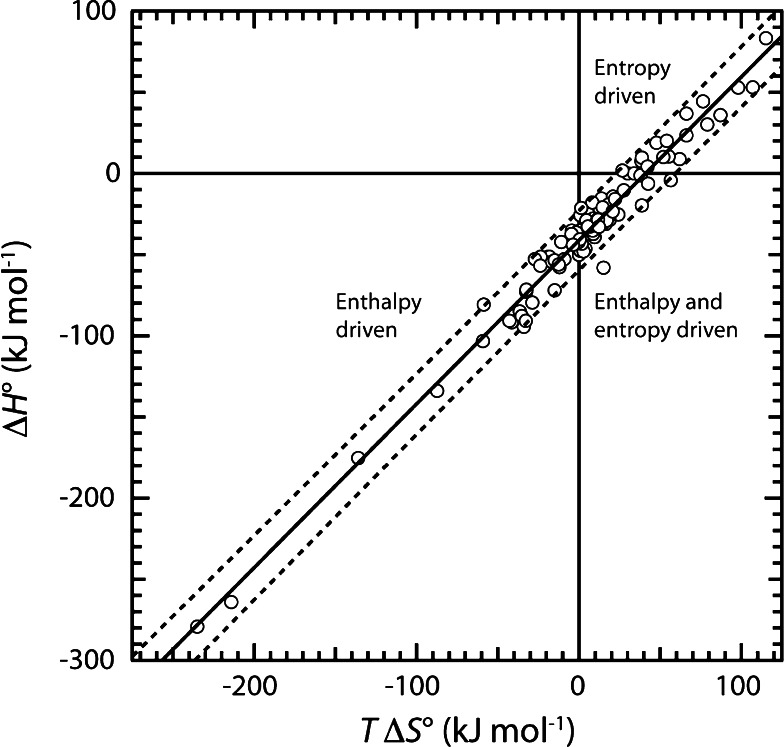
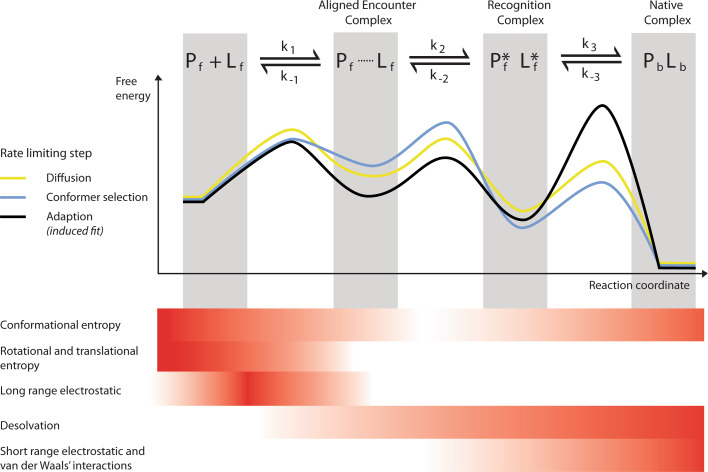
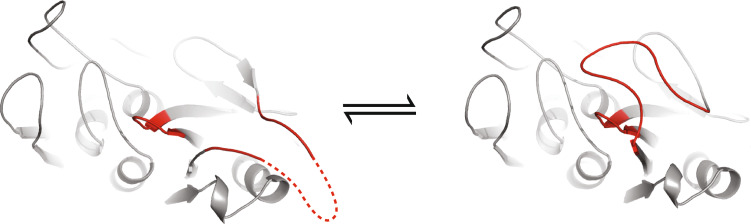
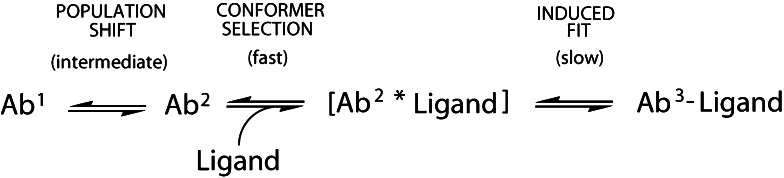
Proteins are dynamic entities, and they possess an inherent flexibility that allows them to function through molecular interactions within the cell, among cells and even between organisms. Appreciation of the non-static nature of proteins is emerging, but to describe and incorporate this into an intuitive perception of protein function is challenging. Flexibility is of overwhelming importance for protein function, and the changes in protein structure during interactions with binding partners can be dramatic. The present review addresses protein flexibility, focusing on protein-ligand interactions. The thermodynamics involved are reviewed, and examples of structure-function studies involving experimentally determined flexibility descriptions are presented. While much remains to be understood about protein flexibility, it is clear that it is encoded within their amino acid sequence and should be viewed as an integral part of their structure.
Figures

References
-
- Maragakis P, Lindorff-Larsen K, Eastwood MP, Dror RO, Klepeis JL, Arkin IT, Jensen MO, Xu H, Trbovic N, Friesner RA, Iii AG, Shaw DE. Microsecond molecular dynamics simulation shows effect of slow loop dynamics on backbone amide order parameters of proteins. J Phys Chem B. 2008;112:6155–6158. doi: 10.1021/jp077018h. - DOI - PMC - PubMed
-
- Linderstrøm-Lang K. Deuterium exchange between peptides and water. Chem Soc Spec Publ. 1955;2:1–20.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
