

Efficient drug lead discovery and optimization
- PMID: 19317443
- PMCID: PMC2727934
- DOI: 10.1021/ar800236t
Efficient drug lead discovery and optimization
Abstract
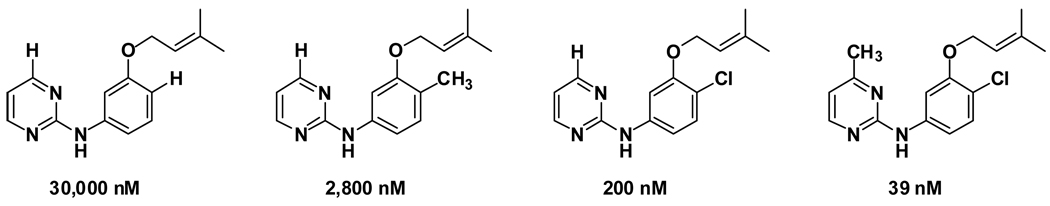
During the 1980s, advances in the abilities to perform computer simulations of chemical and biomolecular systems and to calculate free energy changes led to the expectation that such methodology would soon show great utility for guiding molecular design. Important potential applications included design of selective receptors, catalysts, and regulators of biological function including enzyme inhibitors. This time also saw the rise of high-throughput screening and combinatorial chemistry along with complementary computational methods for de novo design and virtual screening including docking. These technologies appeared poised to deliver diverse lead compounds for any biological target. As with many technological advances, realization of the expectations required significant additional effort and time. However, as summarized here, striking success has now been achieved for computer-aided drug lead generation and optimization. De novo design using both molecular growing and docking are illustrated for lead generation, and lead optimization features free energy perturbation calculations in conjunction with Monte Carlo statistical mechanics simulations for protein-inhibitor complexes in aqueous solution. The specific applications are to the discovery of non-nucleoside inhibitors of HIV reverse transcriptase (HIV-RT) and inhibitors of the binding of the proinflammatory cytokine MIF to its receptor CD74. A standard protocol is presented that includes scans for possible additions of small substituents to a molecular core, interchange of heterocycles, and focused optimization of substituents at one site. Initial leads with activities at low-micromolar concentrations have been advanced rapidly to low-nanomolar inhibitors.
Figures







References
-
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
