

Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint
- PMID: 19322196
- PMCID: PMC2683710
- DOI: 10.1038/emboj.2009.75
Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint
Abstract
The Rad52 pathway has a central function in the recombinational repair of chromosome breaks and in the recovery from replication stress. Tolerance to replication stress also depends on the Mec1 kinase, which activates the DNA replication checkpoint in an Mrc1-dependent manner in response to fork arrest. Although the Mec1 and Rad52 pathways are initiated by the same single-strand DNA (ssDNA) intermediate, their interplay at stalled forks remains largely unexplored. Here, we show that the replication checkpoint suppresses the formation of Rad52 foci in an Mrc1-dependent manner and prevents homologous recombination (HR) at chromosome breaks induced by the HO endonuclease. This repression operates at least in part by impeding resection of DNA ends, which is essential to generate 3' ssDNA tails, the primary substrate of HR. Interestingly, we also observed that the Mec1 pathway does not prevent recombination at stalled forks, presumably because they already contain ssDNA. Taken together, these data indicate that the DNA replication checkpoint suppresses genomic instability in S phase by blocking recombination at chromosome breaks and permitting helpful recombination at stalled forks.
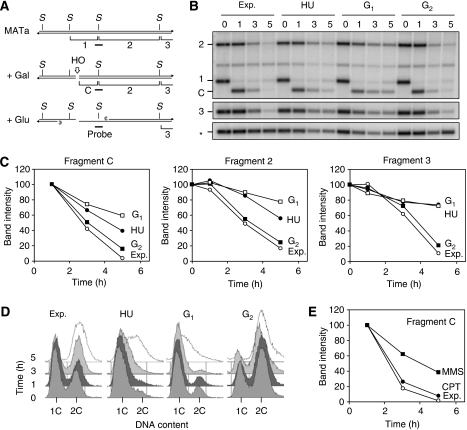
Figures






References
-
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3: 958–965 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
