

Structure and mechanism of protein stability sensors: chaperone activity of small heat shock proteins
- PMID: 19323523
- PMCID: PMC2785012
- DOI: 10.1021/bi900212j
Structure and mechanism of protein stability sensors: chaperone activity of small heat shock proteins
Abstract
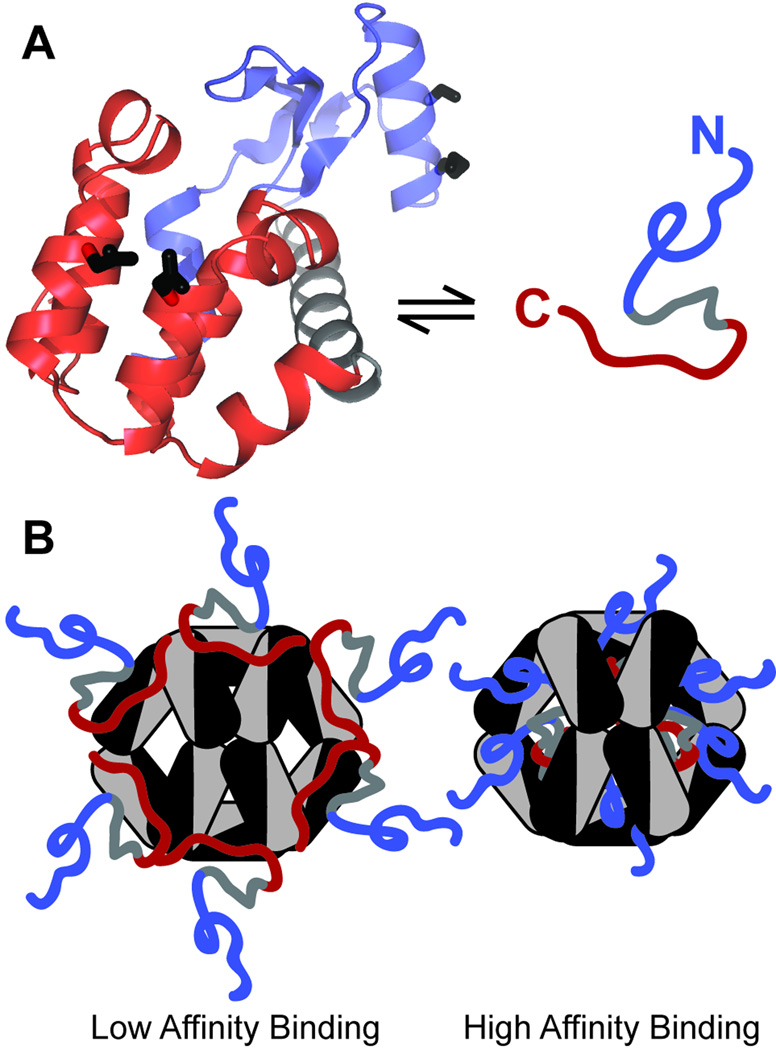
Small heat shock proteins (sHSP) make up a remarkably diverse group of molecular chaperones possessing a degree of structural plasticity unparalleled in other protein superfamilies. In the absence of chemical energy input, these stability sensors can sensitively recognize and bind destabilized proteins, even in the absence of gross misfolding. Cellular conditions regulate affinity toward client proteins, allowing tightly controlled switching and tuning of sHSP chaperone capacity. Perturbations of this regulation, through chemical modification or mutation, directly lead to a variety of disease states. This review explores the structural basis of sHSP oligomeric flexibility and the corresponding functional consequences in the context of a model describing sHSP activity with a set of three coupled thermodynamic equilibria. As current research illuminates many novel physiological roles for sHSP outside of their traditional duties as molecular chaperones, such a conceptual framework provides a sound foundation for describing these emerging functions in physiological and pathological processes.
Figures






References
-
- Dill KA, Chan HS. From Levinthal to pathways to funnels. Nature Structural Biology. 1997;4:10–19. - PubMed
-
- Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annual Review of Physical Chemistry. 1997;48:545–600. - PubMed
-
- Dobson CM. Principles of protein folding, misfolding and aggregation. Seminars in Cell & Developmental Biology. 2004;15:3–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
