

Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein
- PMID: 19325877
- PMCID: PMC2654508
- DOI: 10.1371/journal.ppat.1000353
Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein
Abstract
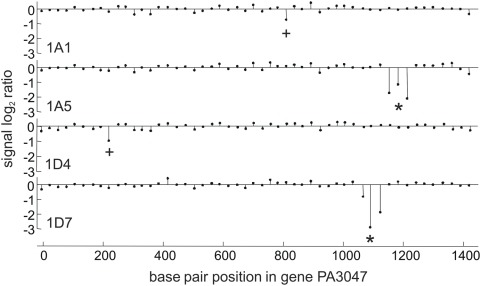
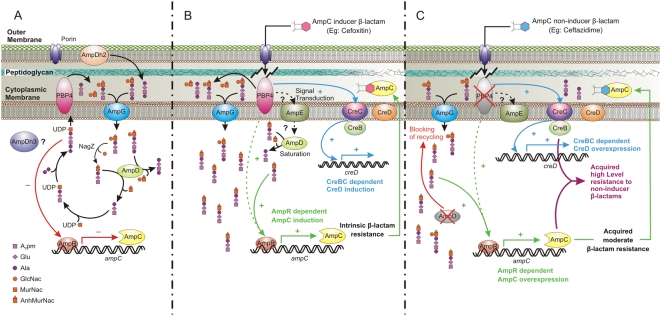
It has long been recognized that the modification of penicillin-binding proteins (PBPs) to reduce their affinity for beta-lactams is an important mechanism (target modification) by which Gram-positive cocci acquire antibiotic resistance. Among Gram-negative rods (GNR), however, this mechanism has been considered unusual, and restricted to clinically irrelevant laboratory mutants for most species. Using as a model Pseudomonas aeruginosa, high up on the list of pathogens causing life-threatening infections in hospitalized patients worldwide, we show that PBPs may also play a major role in beta-lactam resistance in GNR, but through a totally distinct mechanism. Through a detailed genetic investigation, including whole-genome analysis approaches, we demonstrate that high-level (clinical) beta-lactam resistance in vitro, in vivo, and in the clinical setting is driven by the inactivation of the dacB-encoded nonessential PBP4, which behaves as a trap target for beta-lactams. The inactivation of this PBP is shown to determine a highly efficient and complex beta-lactam resistance response, triggering overproduction of the chromosomal beta-lactamase AmpC and the specific activation of the CreBC (BlrAB) two-component regulator, which in turn plays a major role in resistance. These findings are a major step forward in our understanding of beta-lactam resistance biology, and, more importantly, they open up new perspectives on potential antibiotic targets for the treatment of infectious diseases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol Rev. 2008;32:361–385. - PubMed
-
- Vincent JL. Nosocomial infections in adult intensive-care units. Lancet. 2003;361:2068–2077. - PubMed
-
- Livermore DM. Clinical significance of beta-lactamase induction and stable derepression in gram-negative rods. Eur J Clin Microbiol. 1987;6:439–445. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
