

Evaluation of next generation sequencing platforms for population targeted sequencing studies
- PMID: 19327155
- PMCID: PMC2691003
- DOI: 10.1186/gb-2009-10-3-r32
Evaluation of next generation sequencing platforms for population targeted sequencing studies
Abstract
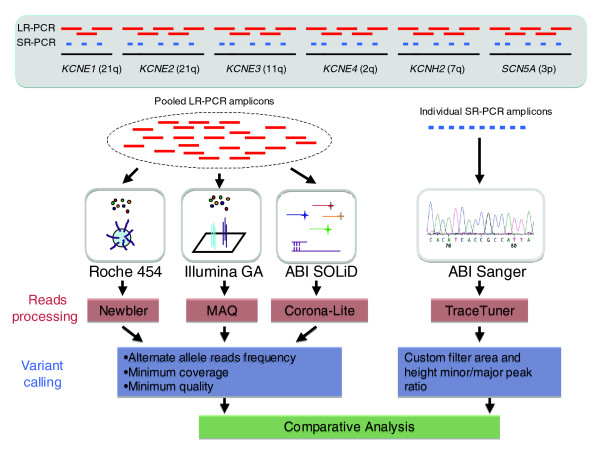
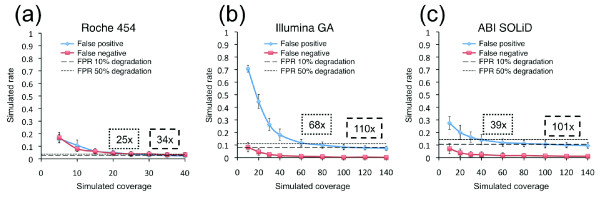
Background: Next generation sequencing (NGS) platforms are currently being utilized for targeted sequencing of candidate genes or genomic intervals to perform sequence-based association studies. To evaluate these platforms for this application, we analyzed human sequence generated by the Roche 454, Illumina GA, and the ABI SOLiD technologies for the same 260 kb in four individuals.
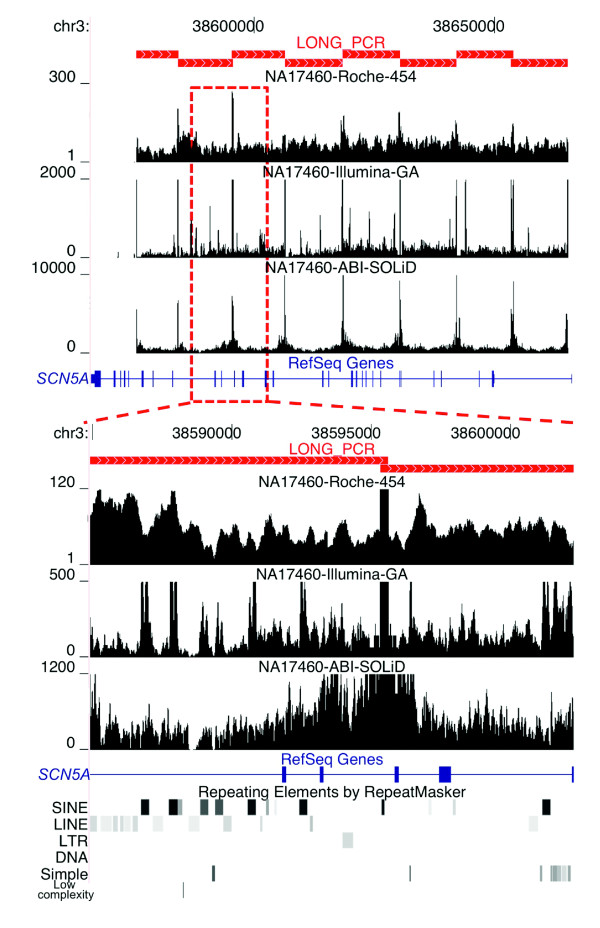
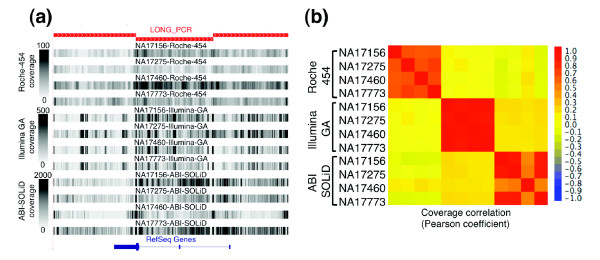
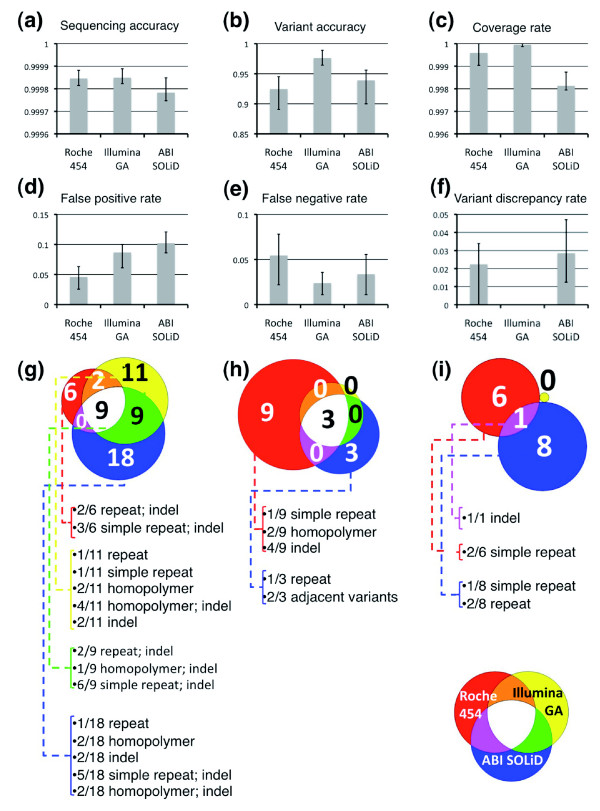
Results: Local sequence characteristics contribute to systematic variability in sequence coverage (>100-fold difference in per-base coverage), resulting in patterns for each NGS technology that are highly correlated between samples. A comparison of the base calls to 88 kb of overlapping ABI 3730xL Sanger sequence generated for the same samples showed that the NGS platforms all have high sensitivity, identifying >95% of variant sites. At high coverage, depth base calling errors are systematic, resulting from local sequence contexts; as the coverage is lowered additional 'random sampling' errors in base calling occur.
Conclusions: Our study provides important insights into systematic biases and data variability that need to be considered when utilizing NGS platforms for population targeted sequencing studies.
Figures
References
-
- Bonetta L. Genome sequencing in the fast lane. Nat Methods. 2006;3:141–147. doi: 10.1038/nmeth0206-141. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
