

Protein structure prediction: when is it useful?
- PMID: 19327982
- PMCID: PMC2673339
- DOI: 10.1016/j.sbi.2009.02.005
Protein structure prediction: when is it useful?
Abstract
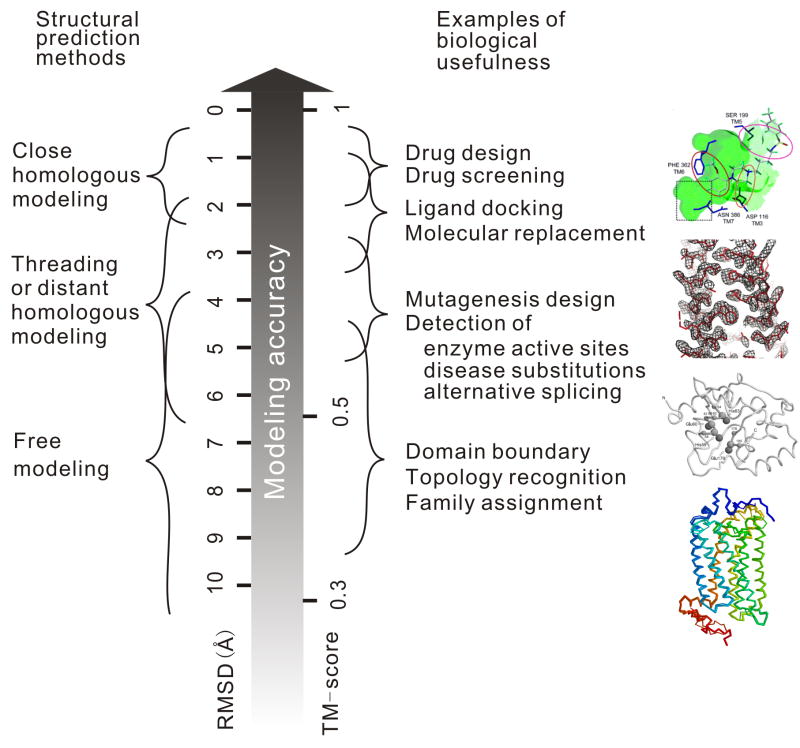
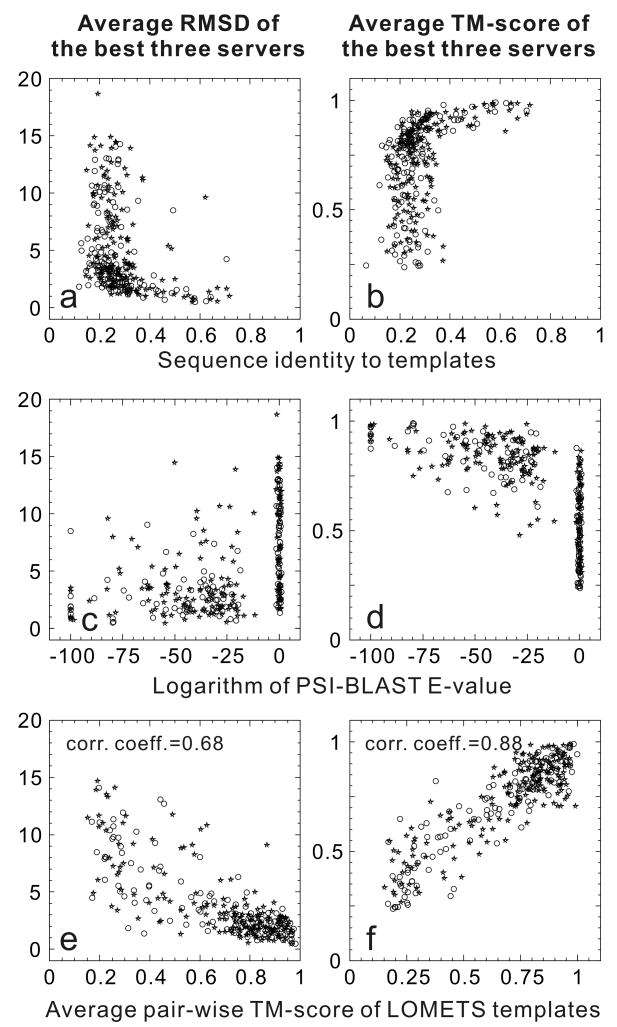
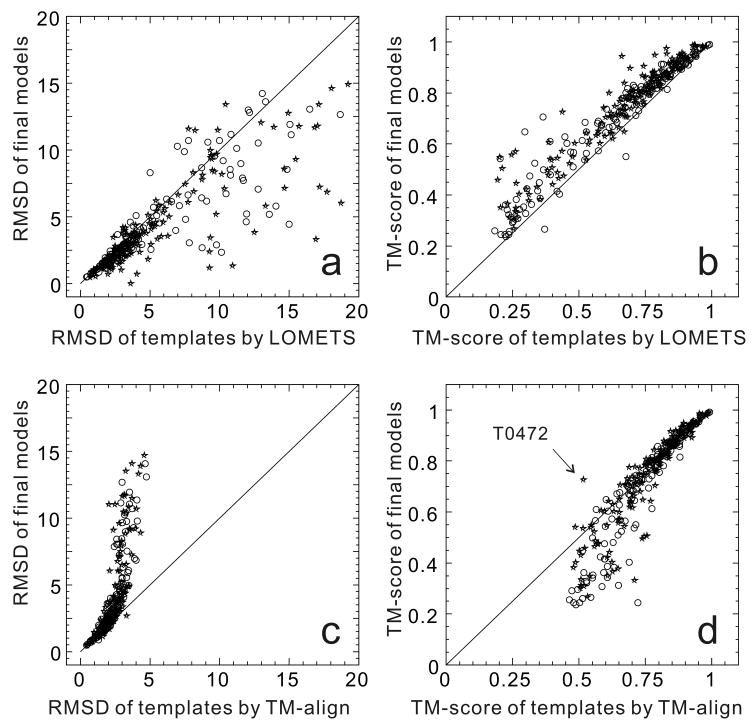
Computationally predicted three-dimensional structure of protein molecules has demonstrated the usefulness in many areas of biomedicine, ranging from approximate family assignments to precise drug screening. For nearly 40 years, however, the accuracy of the predicted models has been dictated by the availability of close structural templates. Progress has recently been achieved in refining low-resolution models closer to the native ones; this has been made possible by combining knowledge-based information from multiple sources of structural templates as well as by improving the energy funnel of physics-based force fields. Unfortunately, there has been no essential progress in the development of techniques for detecting remotely homologous templates and for predicting novel protein structures.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
