

Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation
- PMID: 19332876
- PMCID: PMC2715112
- DOI: 10.1084/jem.20080003
Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation
Abstract
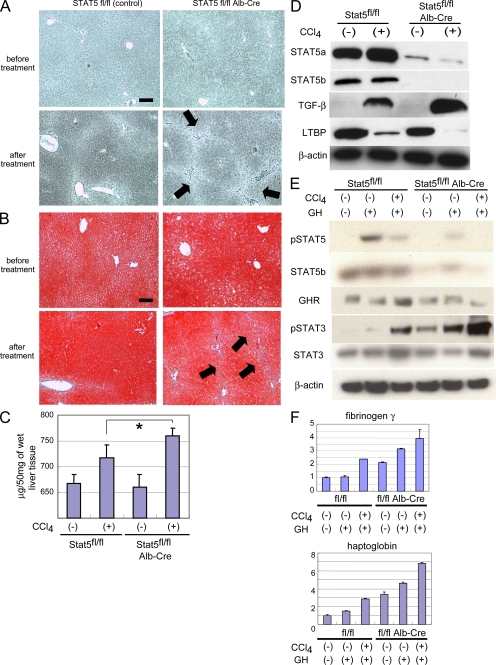
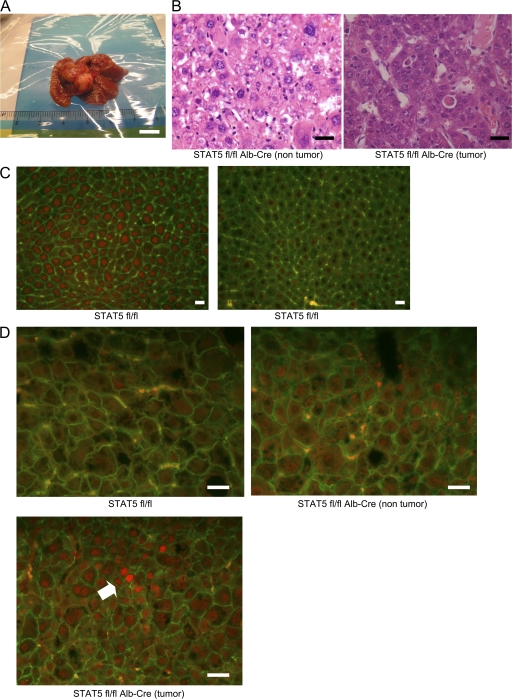
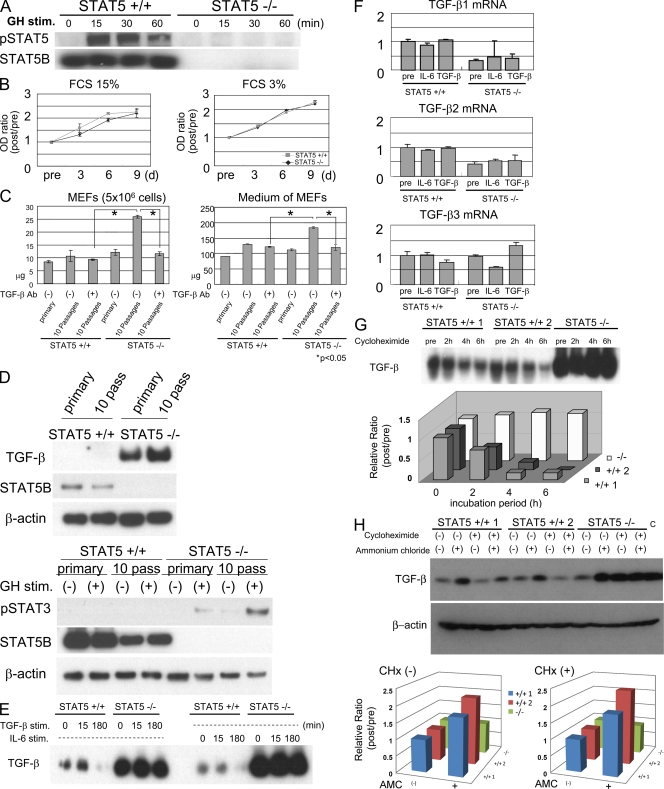
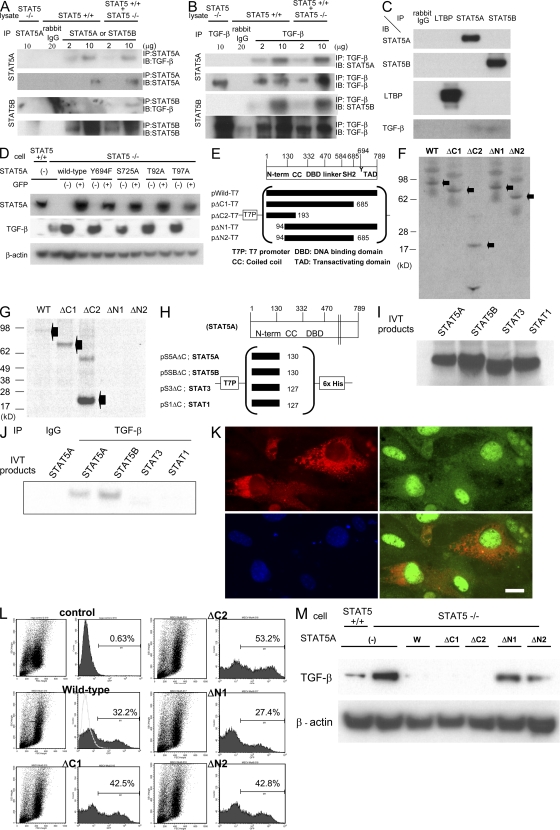
The molecular mechanisms underlying the development of hepatocellular carcinoma are not fully understood. Liver-specific signal transducer and activator of transcription (STAT) 5A/B-null mice (STAT5-LKO) were treated with carbon tetrachloride (CCl(4)), and histological analyses revealed liver fibrosis and tumors. Transforming growth factor (TGF)-beta levels and STAT3 activity were elevated in liver tissue from STAT5-LKO mice upon CCl(4) treatment. To define the molecular link between STAT5 silencing and TGF-beta up-regulation, as well as STAT3 activation, we examined STAT5-null mouse embryonic fibroblasts and primary hepatocytes. These cells displayed elevated TGF-beta protein levels, whereas messenger RNA levels remained almost unchanged. Protease inhibitor studies revealed that STAT5 deficiency enhanced the stability of mature TGF-beta. Immunoprecipitation and immunohistochemistry analyses demonstrated that STAT5, through its N-terminal sequences, could bind to TGF-beta and that retroviral-mediated overexpression of STAT5 decreased TGF-beta levels. To confirm the in vivo significance of the N-terminal domain of STAT5, we treated mice that expressed STAT5 lacking the N terminus (STAT5-DeltaN) with CCl(4). STAT5-DeltaN mice developed CCl(4)-induced liver fibrosis but no tumors. In conclusion, loss of STAT5 results in elevated TGF-beta levels and enhanced growth hormone-induced STAT3 activity. We propose that a deregulated STAT5-TGF-beta-STAT3 network contributes to the development of chronic liver disease.
Figures
References
-
- Yoshikawa H., Matsubara K., Qian G.S., Jackson P., Groopman J.D., Manning J.E., Harris C.C., Herman J.G. 2001. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity.Nat. Genet. 28:29–35 - PubMed
-
- Satoh S., Daigo Y., Furukawa Y., Kato T., Miwa N., Nishiwaki T., Kawasoe T., Ishiguro H., Fujita M., Tokino T., et al. 2000. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1.Nat. Genet. 24:245–250 - PubMed
-
- Derynck R., Jarrett J.A., Chen E.Y., Eaton D.H., Bell J.R., Assoian R.K., Roberts A.B., Sporn M.B., Goeddel D.V. 1985. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells.Nature. 316:701–705 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
