

Lysosomal storage disorders in the newborn
- PMID: 19336380
- PMCID: PMC2768319
- DOI: 10.1542/peds.2008-0635
Lysosomal storage disorders in the newborn
Abstract
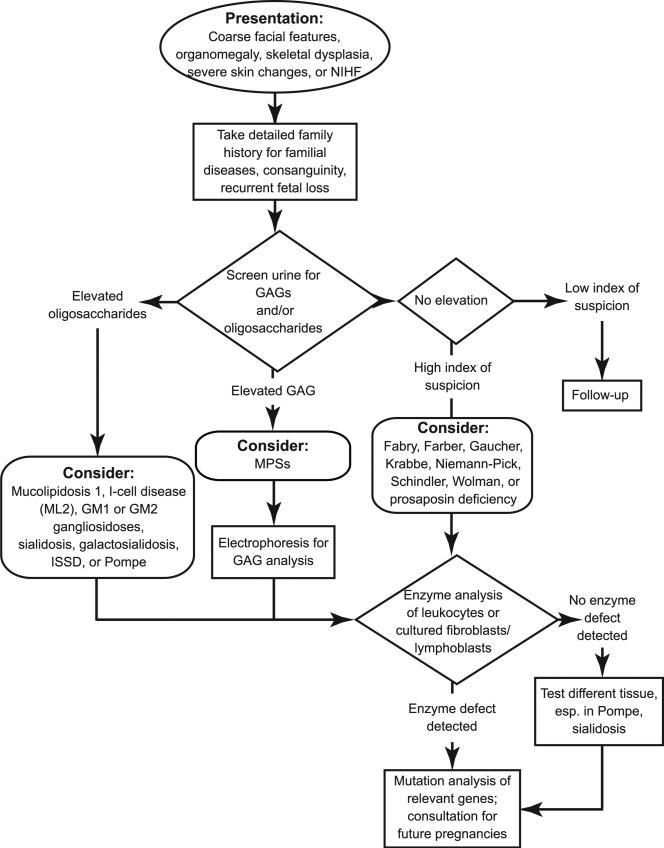
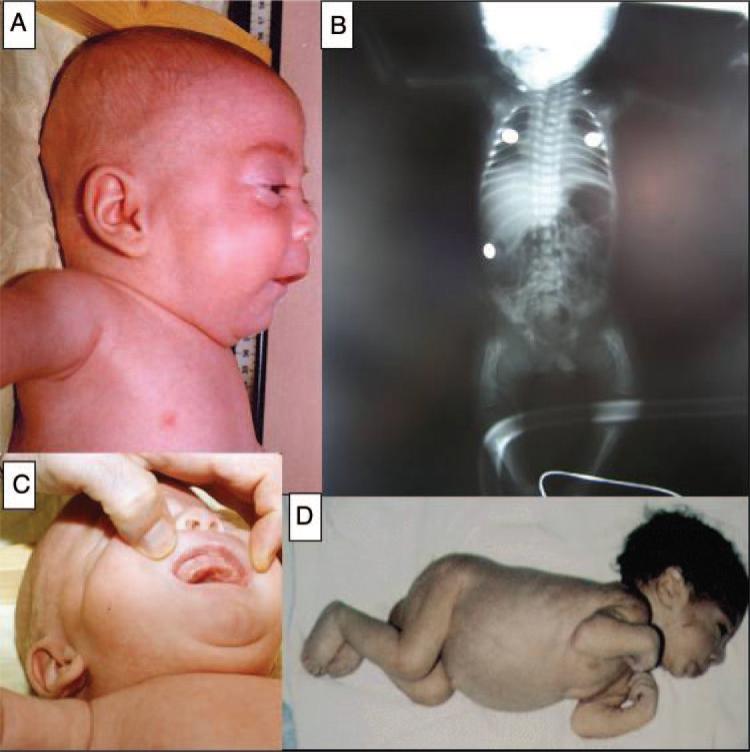
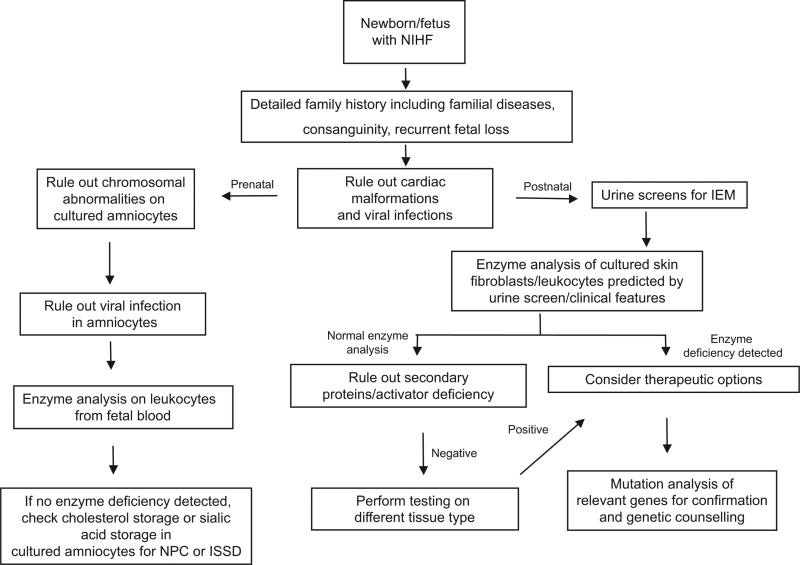
Lysosomal storage disorders are rare inborn errors of metabolism, with a combined incidence of 1 in 1500 to 7000 live births. These relatively rare disorders are seldom considered when evaluating a sick newborn. A significant number of the >50 different lysosomal storage disorders, however, do manifest in the neonatal period and should be part of the differential diagnosis of several perinatal phenotypes. We review the earliest clinical features, diagnostic tests, and treatment options for lysosomal storage disorders that can present in the newborn. Although many of the lysosomal storage disorders are characterized by a range in phenotypes, the focus of this review is on the specific symptoms and clinical findings that present in the perinatal period, including neurologic, respiratory, endocrine, and cardiovascular manifestations, dysmorphic features, hepatosplenomegaly, skin or ocular involvement, and hydrops fetalis/congenital ascites. A greater awareness of these features may help to reduce misdiagnosis and promote the early detection of lysosomal storage disorders. Implementing therapy at the earliest stage possible is crucial for several of the lysosomal storage disorders; hence, an early appreciation of these disorders by physicians who treat newborns is essential.
Figures
References
-
- Stone DL, Sidransky E. Hydrops fetalis: lysosomal storage disorders in extremis. Adv Pediatr. 1999;46:409–440. - PubMed
-
- Fletcher JM. Screening for lysosomal storage disorders: a clinical perspective. J Inherit Metab Dis. 2006;29(2–3):405–408. - PubMed
-
- Beaudet AL, Scriver CR, Sly WS, Valle D. Genetics, biochemistry and molecular basis of variant human phenotypes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. McGraw-Hill; New York, NY: 1995. pp. 53–118.
-
- Wraith JE. Lysosomal disorders. Semin Neonatol. 2002;7(1):75–83. - PubMed
-
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5(7):554–565. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
