

Vascular consequences of dietary salt intake
- PMID: 19339634
- PMCID: PMC2724242
- DOI: 10.1152/ajprenal.00027.2009
Vascular consequences of dietary salt intake
Abstract
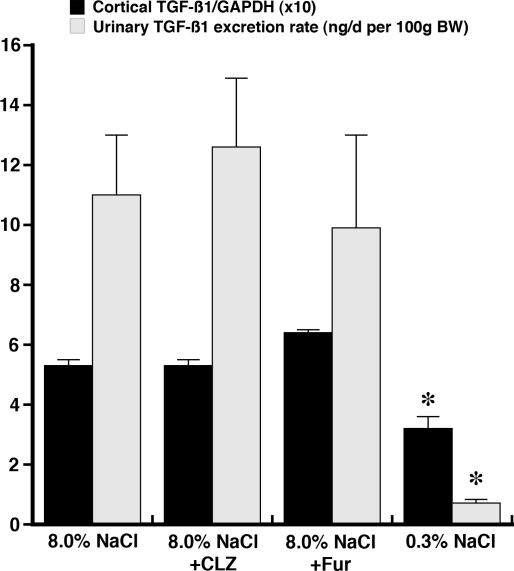
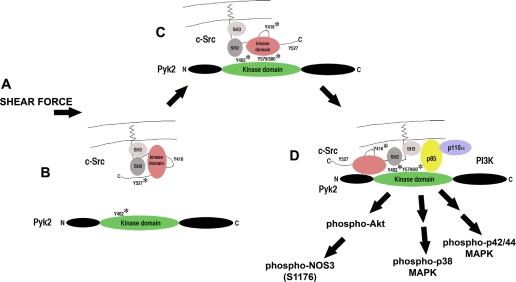
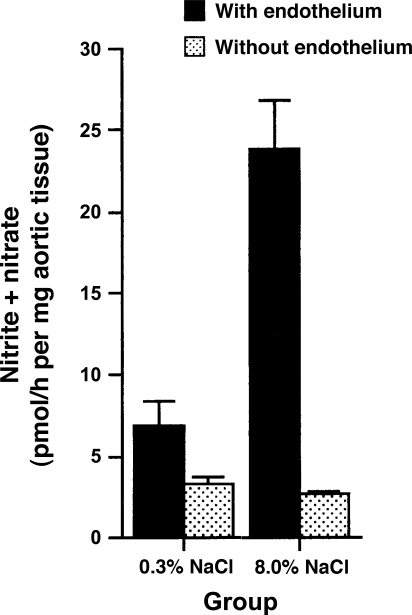
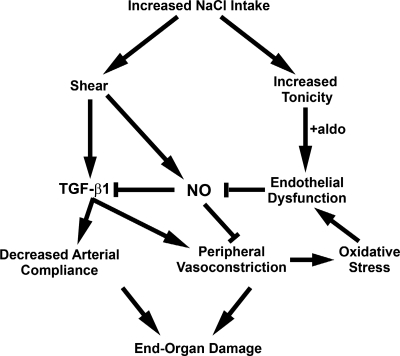
Animal and human studies support an untoward effect of excess dietary NaCl (salt) intake on cardiovascular and renal function and life span. Recent work has promoted the concept that the endothelium, in particular, reacts to changes in dietary salt intake through a complex series of events that are independent of blood pressure and the renin-angiotensin-aldosterone axis. The cellular signaling events culminate in the intravascular production of transforming growth factor-beta (TGF-beta) and nitric oxide in response to increased salt intake. Plasticity of the endothelium is integral in the vascular remodeling consequences associated with excess salt intake, because nitric oxide serves as a negative regulator of TGF-beta production. Impairment of nitric oxide production, such as occurs with endothelial dysfunction in a variety of disease states, results in unopposed excess vascular TGF-beta production, which promotes reduced vascular compliance and augmented peripheral arterial constriction and hypertension. Persistent alterations in vascular function promote the increase in cardiovascular events and reductions in renal function that reduce life span during increased salt intake.
Figures

References
-
- Akagi Y, Isaka Y, Arai M, Kaneko T, Takenaka M, Moriyama T, Kaneda Y, Ando A, Orita Y, Kamada T, Ueda N, Imai E. Inhibition of TGF-β1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int 50: 148–155, 1996. - PubMed
-
- Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 24: 413–420, 2004. - PubMed
-
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci 116: 217–224, 2003. - PubMed
-
- Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal 12: 123–133, 2000. - PubMed
-
- Azuma N, Akasaka N, Kito H, Ikeda M, Gahtan V, Sasajima T, Sumpio BE. Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress. Am J Physiol Heart Circ Physiol 280: H189–H197, 2001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical