

Mouse models of human AML accurately predict chemotherapy response
- PMID: 19339691
- PMCID: PMC2666344
- DOI: 10.1101/gad.1771409
Mouse models of human AML accurately predict chemotherapy response
Abstract
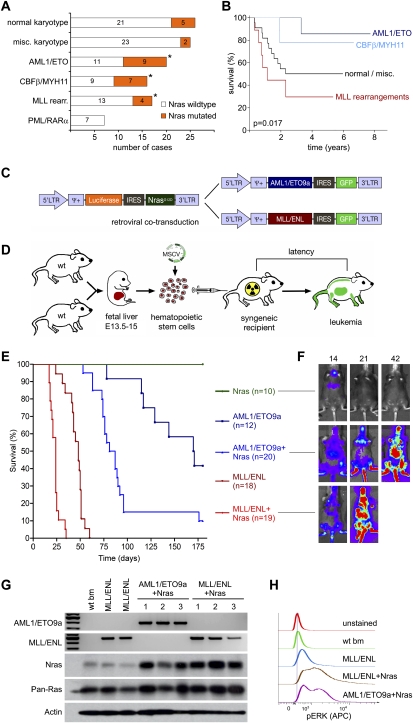
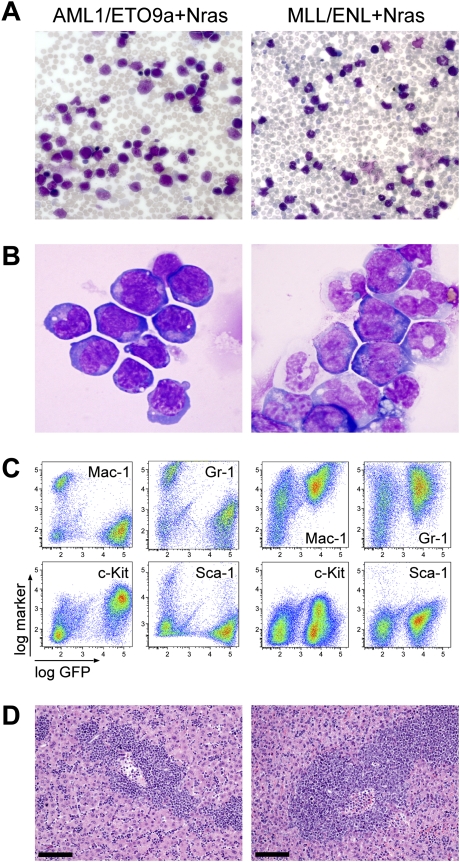
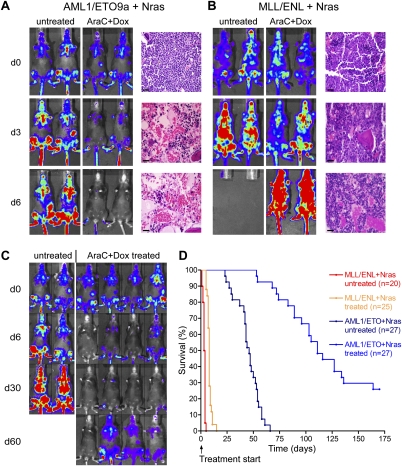
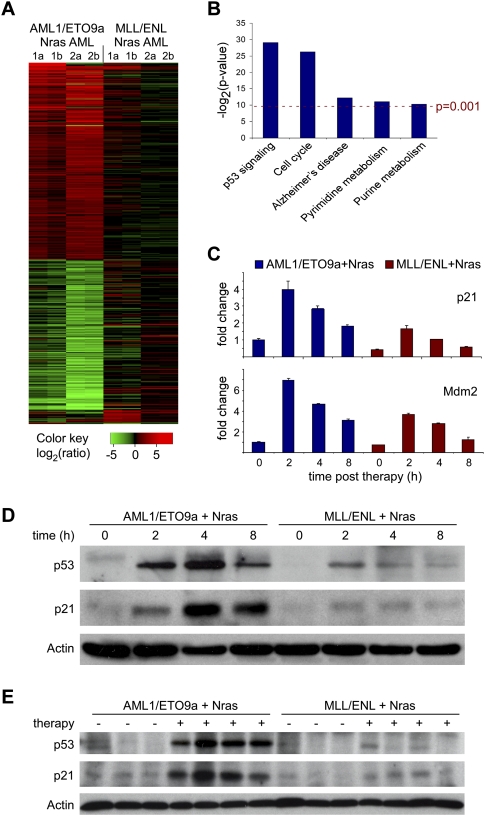
The genetic heterogeneity of cancer influences the trajectory of tumor progression and may underlie clinical variation in therapy response. To model such heterogeneity, we produced genetically and pathologically accurate mouse models of common forms of human acute myeloid leukemia (AML) and developed methods to mimic standard induction chemotherapy and efficiently monitor therapy response. We see that murine AMLs harboring two common human AML genotypes show remarkably diverse responses to conventional therapy that mirror clinical experience. Specifically, murine leukemias expressing the AML1/ETO fusion oncoprotein, associated with a favorable prognosis in patients, show a dramatic response to induction chemotherapy owing to robust activation of the p53 tumor suppressor network. Conversely, murine leukemias expressing MLL fusion proteins, associated with a dismal prognosis in patients, are drug-resistant due to an attenuated p53 response. Our studies highlight the importance of genetic information in guiding the treatment of human AML, functionally establish the p53 network as a central determinant of chemotherapy response in AML, and demonstrate that genetically engineered mouse models of human cancer can accurately predict therapy response in patients.
Figures
References
-
- Anensen N., Oyan A.M., Bourdon J.C., Kalland K.H., Bruserud O., Gjertsen B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. 2006;12:3985–3992. - PubMed
-
- Byrd J.C., Mrozek K., Dodge R.K., Carroll A.J., Edwards C.G., Arthur D.C., Pettenati M.J., Patil S.R., Rao K.W., Watson M.S., et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials