

Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation
- PMID: 19340588
- PMCID: PMC2693357
- DOI: 10.1007/s10822-009-9266-3
Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation
Abstract
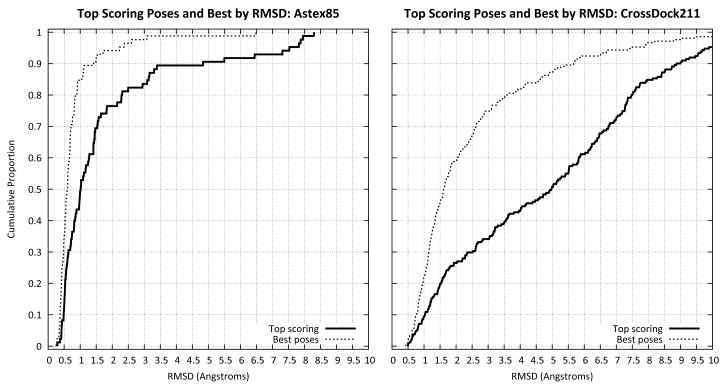
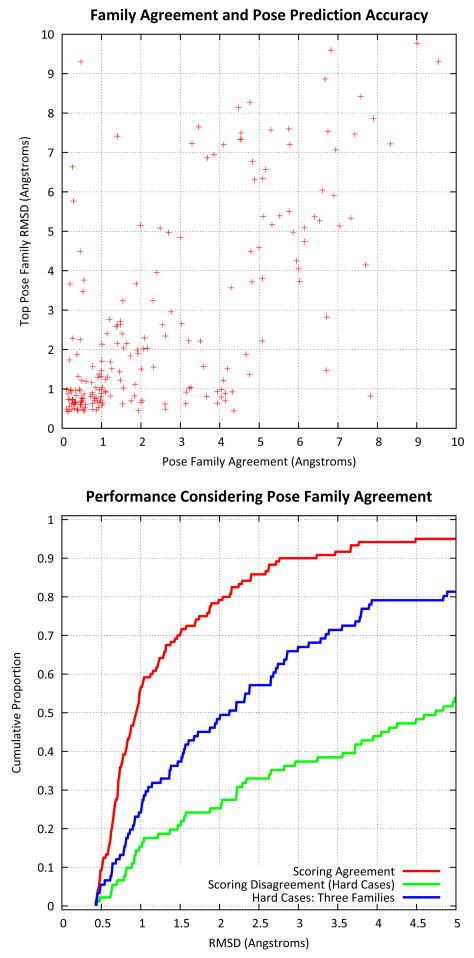
Computational methods for docking ligands have been shown to be remarkably dependent on precise protein conformation, where acceptable results in pose prediction have been generally possible only in the artificial case of re-docking a ligand into a protein binding site whose conformation was determined in the presence of the same ligand (the "cognate" docking problem). In such cases, on well curated protein/ligand complexes, accurate dockings can be returned as top-scoring over 75% of the time using tools such as Surflex-Dock. A critical application of docking in modeling for lead optimization requires accurate pose prediction for novel ligands, ranging from simple synthetic analogs to very different molecular scaffolds. Typical results for widely used programs in the "cross-docking case" (making use of a single fixed protein conformation) have rates closer to 20% success. By making use of protein conformations from multiple complexes, Surflex-Dock yields an average success rate of 61% across eight pharmaceutically relevant targets. Following docking, protein pocket adaptation and rescoring identifies single pose families that are correct an average of 67% of the time. Consideration of the best of two pose families (from alternate scoring regimes) yields a 75% mean success rate.
Figures











References
-
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161(2):269–88. - PubMed
-
- Welch W, Ruppert J, Jain AN. Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol. 1996;3(6):449–62. - PubMed
-
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261(3):470–89. - PubMed
-
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–48. - PubMed
-
- Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9(1):1–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
