

Pegylated leptin antagonist is a potent orexigenic agent: preparation and mechanism of activity
- PMID: 19342450
- PMCID: PMC2703547
- DOI: 10.1210/en.2008-1706
Pegylated leptin antagonist is a potent orexigenic agent: preparation and mechanism of activity
Abstract
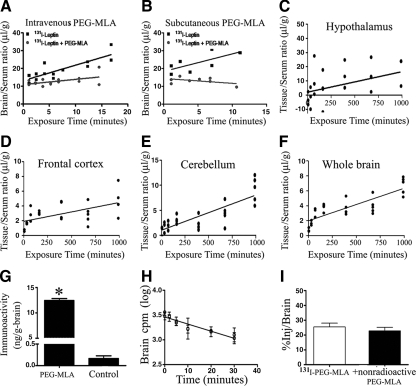
Leptin, a pleiotropic adipokine, is a central regulator of appetite and weight and a key immunomodulatory protein. Although inborn leptin deficiency causes weight gain, it is unclear whether induced leptin deficiency in adult wild-type animals would be orexigenic. Previous work with a potent competitive leptin antagonist did not induce a true metabolic state of leptin deficiency in mice because of a short circulating half-life. In this study, we increased the half-life of the leptin antagonist by pegylation, which resulted in significantly increased bioavailability and retaining of antagonistic activity. Mice administered the pegylated antagonist showed a rapid and dramatic increase in food intake with weight gain. Resulting fat was confined to the mesenteric region with no accumulation in the liver. Serum cholesterol, triglyceride, and hepatic aminotransferases remained unaffected. Weight changes were reversible on cessation of leptin antagonist treatment. The mechanism of severe central leptin deficiency was found to be primarily caused by blockade of transport of circulating leptin across the blood-brain barrier with antagonisms at the arcuate nucleus playing a more minor role. Altogether we introduce a novel compound that induces central and peripheral leptin deficiency. This compound should be useful in exploring the involvement of leptin in metabolic and immune processes and could serve as a therapeutic for the treatment of cachexia.
Figures
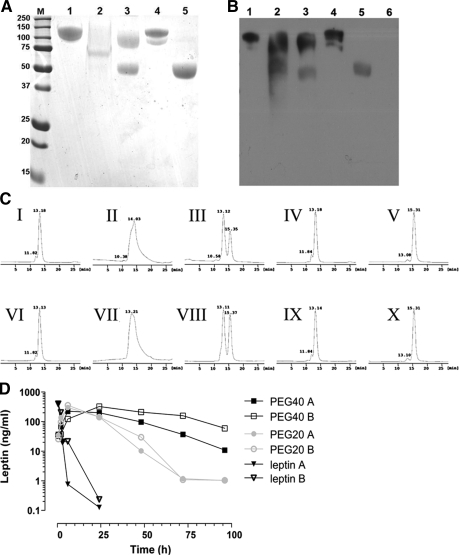
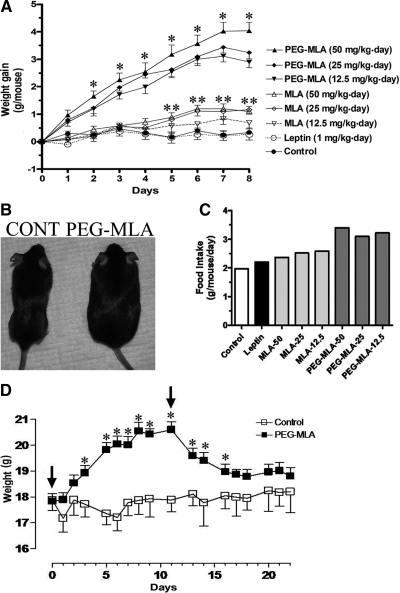
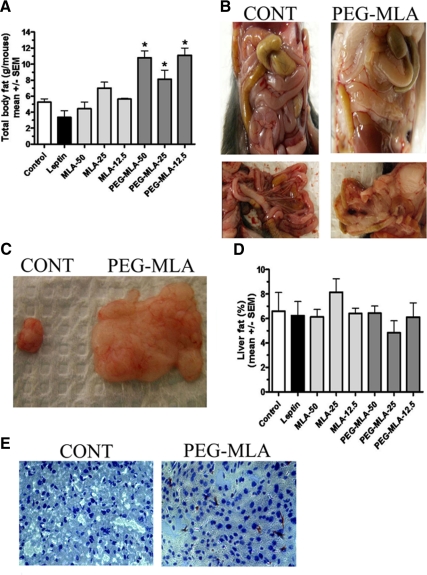

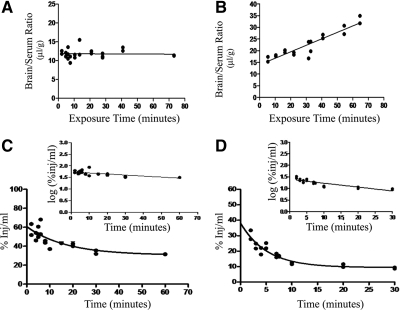
References
-
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM 1994 Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 - PubMed
-
- Karmazyn M, Purdham DM, Rajapurohitam V, Zeidan A 2007 Leptin as a cardiac hypertrophic factor: a potential target for therapeutics. Trends Cardiovasc Med 17:206–211 - PubMed
-
- Peelman F, Waelput W, Iserentant H, Lavens D, Eyckerman S, Zabeau L, Tavernier J 2004 Leptin: linking adipocyte metabolism with cardiovascular and autoimmune diseases. Prog Lipid Res 43:283–301 - PubMed
-
- La Cava A, Matarese G 2004 The weight of leptin in immunity. Nat Rev Immunol 4:371–379 - PubMed
-
- Matarese G, Moschos S, Mantzoros CS 2005 Leptin in immunology. J Immunol 174:3137–3142 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
