

Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study
- PMID: 19345147
- PMCID: PMC3712754
- DOI: 10.1016/S1474-4422(09)70081-X
Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study
Abstract
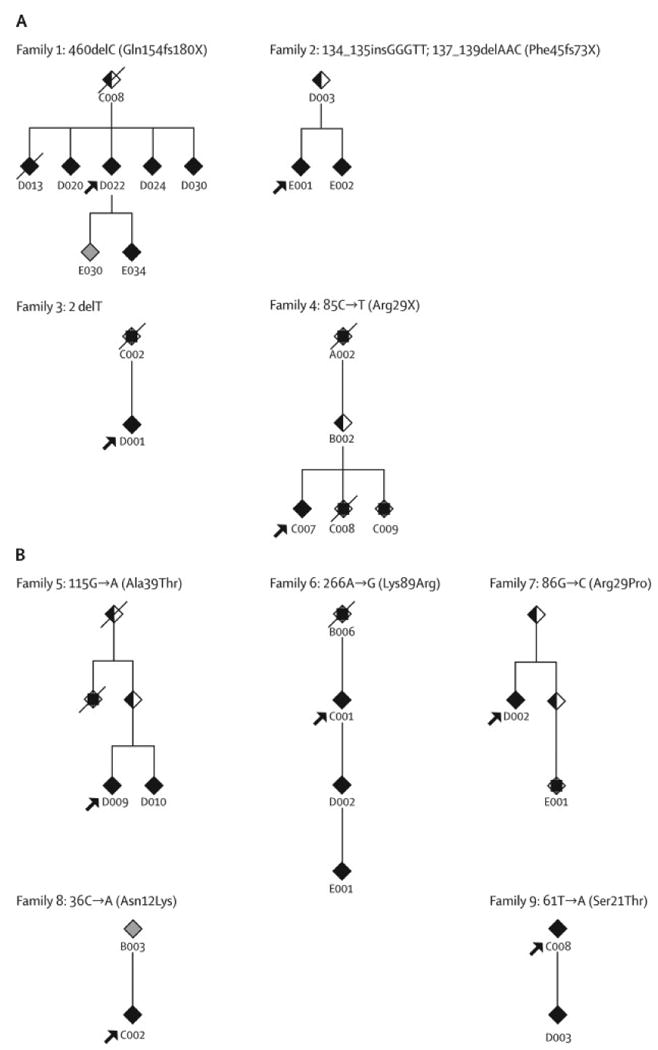
Background: Mutations in THAP1 were recently identified as the cause of DYT6 primary dystonia; a founder mutation was detected in Amish-Mennonite families, and a different mutation was identified in another family of European descent. To assess more broadly the role of this gene, we screened for mutations in families that included one family member who had early-onset, non-focal primary dystonia.
Methods: We identified 36 non-DYT1 multiplex families in which at least one person had non-focal involvement at an age of onset that was younger than 22 years. All three coding exons of THAP1 were sequenced, and the clinical features of individuals with mutations were compared with those of individuals who were negative for mutations in THAP1. Genotype-phenotype differences were also assessed.
Findings: Of 36 families, nine (25%) had members with mutations in THAP1, and most were of German, Irish, or Italian ancestry. One family had the Amish-Mennonite founder mutation, whereas the other eight families each had novel, potentially truncating or missense mutations. The clinical features of the families with mutations conformed to the previously described DYT6 phenotype; however, age at onset was extended from 38 years to 49 years. Compared with non-carriers, mutation carriers were younger at onset and their dystonia was more likely to begin in brachial, rather than cervical, muscles, become generalised, and include speech involvement. Genotype-phenotype differences were not found.
Interpretation: Mutations in THAP1 underlie a substantial proportion of early-onset primary dystonia in non-DYT1 families. The clinical features that are characteristic of affected individuals who have mutations in THAP1 include limb and cranial muscle involvement, and speech is often affected.
Funding: Dystonia Medical Research Foundation; Bachmann-Strauss Dystonia and Parkinson Foundation; National Institute of Neurological Disorders and Stroke; Aaron Aronov Family Foundation.
Conflict of interest statement
Figures

Comment in
-
Transcriptional dysregulation: a cause of dystonia?Lancet Neurol. 2009 May;8(5):416-8. doi: 10.1016/S1474-4422(09)70087-0. Epub 2009 Apr 1. Lancet Neurol. 2009. PMID: 19345149 No abstract available.
References
-
- Ozelius LJ, Bressman SB. DYT1 dystonia. In: Warner TT, Bressman SB, editors. Clinical Diagnosis and Management of Dystonia. London: Informa healthcare; 2007. pp. 53–64.
-
- Bressman SB. Genetics of dystonia: an overview. Parkinsonism Relat Disord. 2007;13(3):S347–355. - PubMed
-
- Chouery E, Kfoury J, Delaugue V, et al. A novel locus for autosomal recessive primary torsion dystonia (DYT17) maps to 20p11.22-q13.12. Neurogenetics. 2008;9:287–293. - PubMed
-
- Fuchs T, Saunders-Pullman R, Raymond D, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet Epub. 2009 Feb 1; - PubMed
-
- Bressman SB, Sabatti C, Raymond D, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2000;54:1746–1752. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
