

Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p
- PMID: 19346491
- PMCID: PMC2672541
- DOI: 10.1073/pnas.0811091106
Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p
Abstract
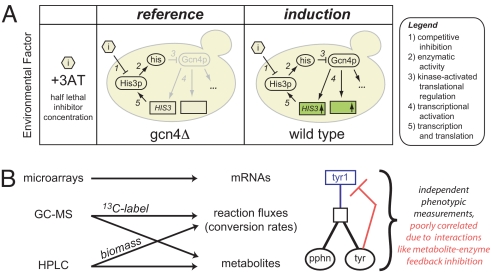
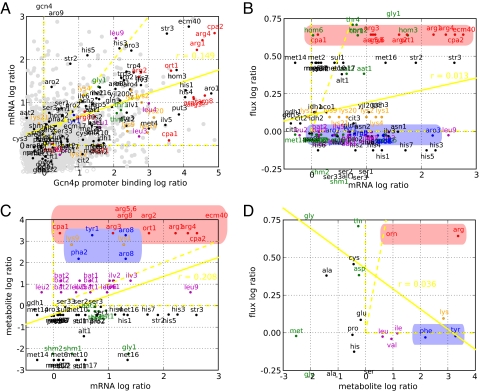
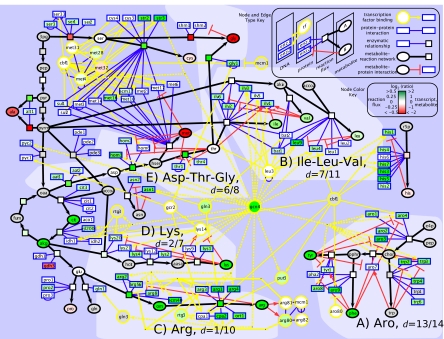
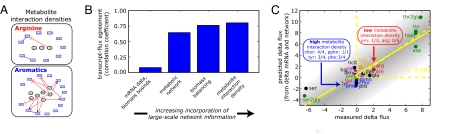
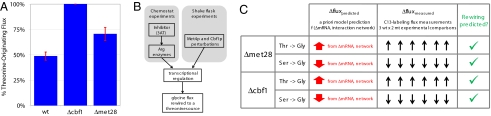
Genome sequencing dramatically increased our ability to understand cellular response to perturbation. Integrating system-wide measurements such as gene expression with networks of protein-protein interactions and transcription factor binding revealed critical insights into cellular behavior. However, the potential of systems biology approaches is limited by difficulties in integrating metabolic measurements across the functional levels of the cell despite their being most closely linked to cellular phenotype. To address this limitation, we developed a model-based approach to correlate mRNA and metabolic flux data that combines information from both interaction network models and flux determination models. We started by quantifying 5,764 mRNAs, 54 metabolites, and 83 experimental (13)C-based reaction fluxes in continuous cultures of yeast under stress in the absence or presence of global regulator Gcn4p. Although mRNA expression alone did not directly predict metabolic response, this correlation improved through incorporating a network-based model of amino acid biosynthesis (from r = 0.07 to 0.80 for mRNA-flux agreement). The model provides evidence of general biological principles: rewiring of metabolic flux (i.e., use of different reaction pathways) by transcriptional regulation and metabolite interaction density (i.e., level of pairwise metabolite-protein interactions) as a key biosynthetic control determinant. Furthermore, this model predicted flux rewiring in studies of follow-on transcriptional regulators that were experimentally validated with additional (13)C-based flux measurements. As a first step in linking metabolic control and genetic regulatory networks, this model underscores the importance of integrating diverse data types in large-scale cellular models. We anticipate that an integrated approach focusing on metabolic measurements will facilitate construction of more realistic models of cellular regulation for understanding diseases and constructing strains for industrial applications.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Uetz P, et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. - PubMed
-
- Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. - PubMed
-
- Ideker T, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. - PubMed
-
- Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl 1):S233–240. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
