

Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF)
- PMID: 19347046
- PMCID: PMC2661376
- DOI: 10.1371/journal.pone.0005134
Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF)
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a progressive, chronic interstitial lung disease that is unresponsive to current therapy and often leads to death. However, the rate of disease progression differs among patients. We hypothesized that comparing the gene expression profiles between patients with stable disease and those in which the disease progressed rapidly will lead to biomarker discovery and contribute to the understanding of disease pathogenesis.
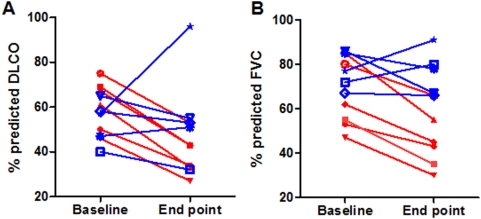
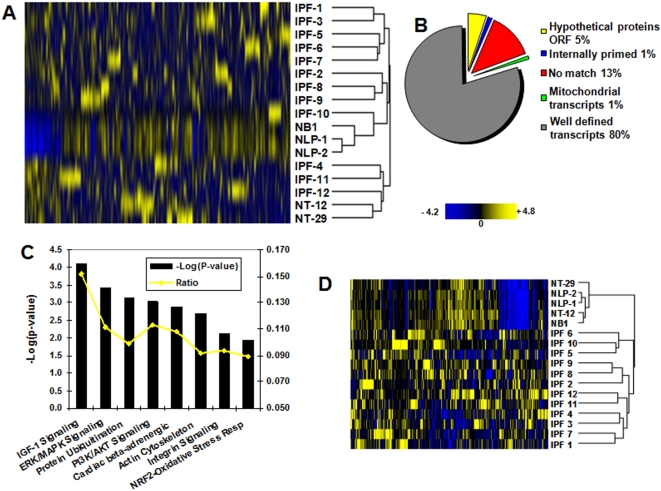
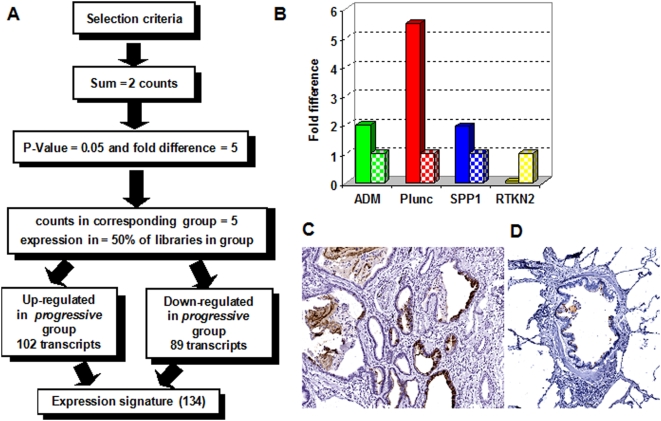
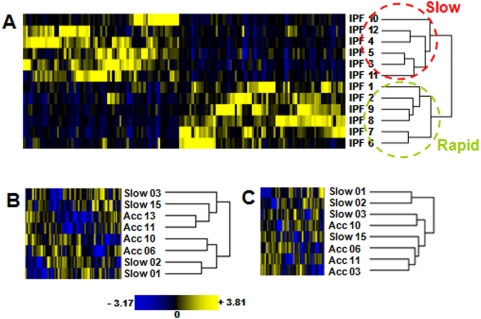
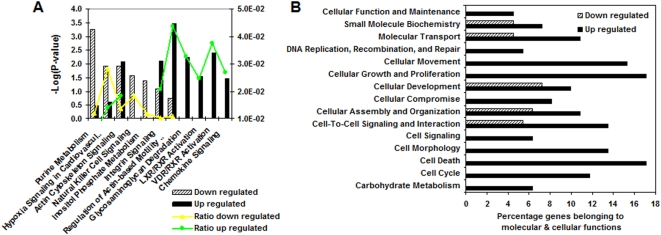
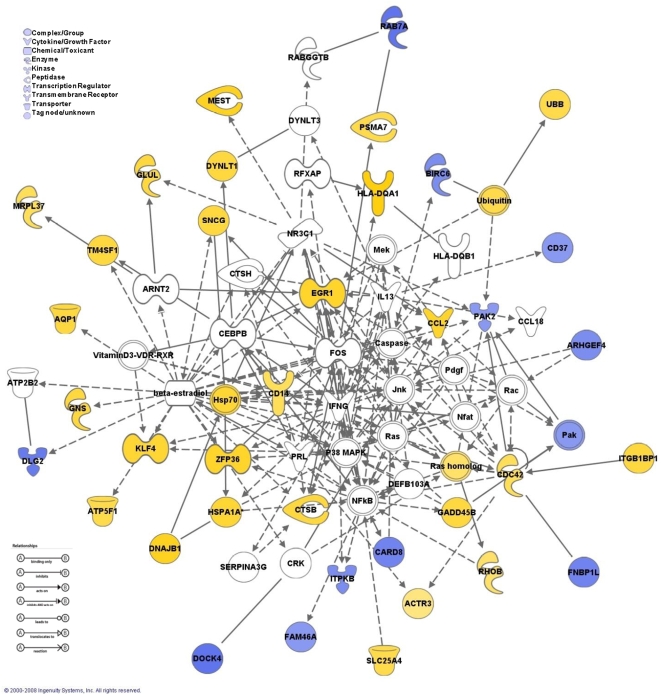
Methodology and principal findings: To begin to address this hypothesis, we applied Serial Analysis of Gene Expression (SAGE) to generate lung expression profiles from diagnostic surgical lung biopsies in 6 individuals with relatively stable (or slowly progressive) IPF and 6 individuals with progressive IPF (based on changes in DLCO and FVC over 12 months). Our results indicate that this comprehensive lung IPF SAGE transcriptome is distinct from normal lung tissue and other chronic lung diseases. To identify candidate markers of disease progression, we compared the IPF SAGE profiles in stable and progressive disease, and identified a set of 102 transcripts that were at least 5-fold up regulated and a set of 89 transcripts that were at least 5-fold down regulated in the progressive group (P-value</=0.05). The over expressed genes included surfactant protein A1, two members of the MAPK-EGR-1-HSP70 pathway that regulate cigarette-smoke induced inflammation, and Plunc (palate, lung and nasal epithelium associated), a gene not previously implicated in IPF. Interestingly, 26 of the up regulated genes are also increased in lung adenocarcinomas and have low or no expression in normal lung tissue. More importantly, we defined a SAGE molecular expression signature of 134 transcripts that sufficiently distinguished relatively stable from progressive IPF.
Conclusions: These findings indicate that molecular signatures from lung parenchyma at the time of diagnosis could prove helpful in predicting the likelihood of disease progression or possibly understanding the biological activity of IPF.
Conflict of interest statement
Figures
References
-
- ATS Dyspnea. Mechanisms, Assessment, and Management: A Consensus Statement. Am J Respir Crit Care Med. 1999;159:321–340. - PubMed
-
- ATS American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This Joint Statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS Board of Directors, June 2001 and by The ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. - PubMed
-
- Gharaee-Kermani M, Gyetko M, Hu B, Phan S. New Insights into the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis: A Potential Role for Stem Cells in the Lung Parenchyma and Implications for Therapy. Pharmaceutical Research. 2007;24:819–841. - PubMed
-
- Maher TM, Wells AU, Laurent GJ. Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J. 2007;30:835–839. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
