

Mechanism and energetics of charybdotoxin unbinding from a potassium channel from molecular dynamics simulations
- PMID: 19348743
- PMCID: PMC2711292
- DOI: 10.1016/j.bpj.2008.12.3952
Mechanism and energetics of charybdotoxin unbinding from a potassium channel from molecular dynamics simulations
Abstract
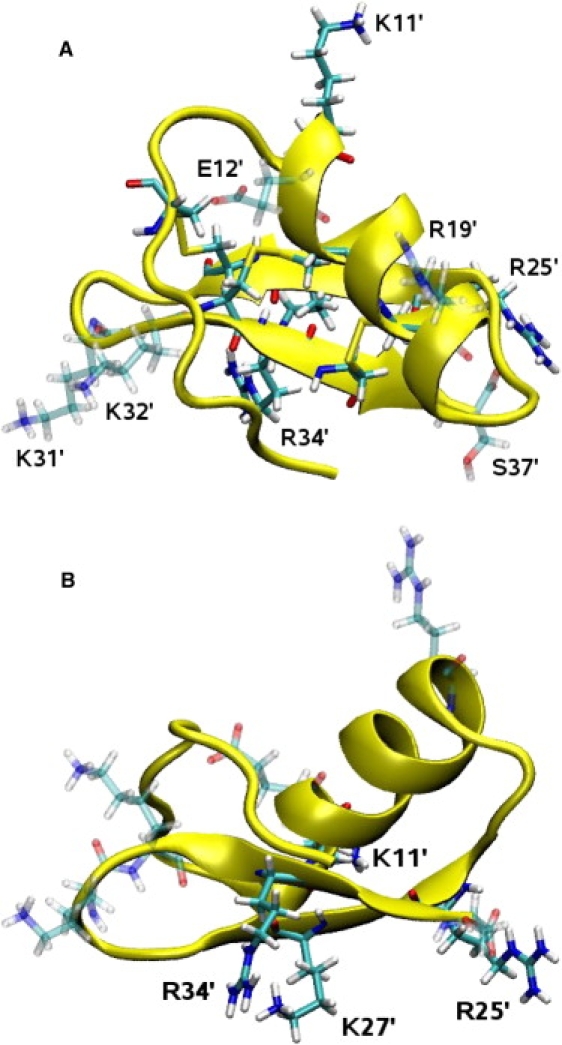
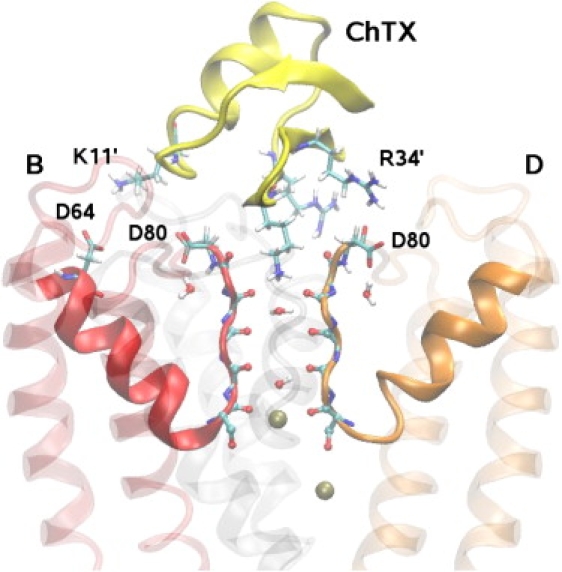
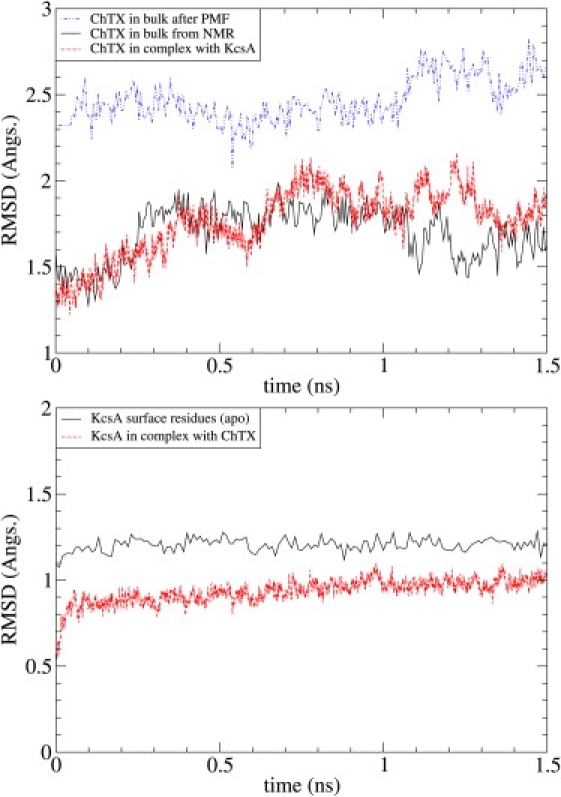
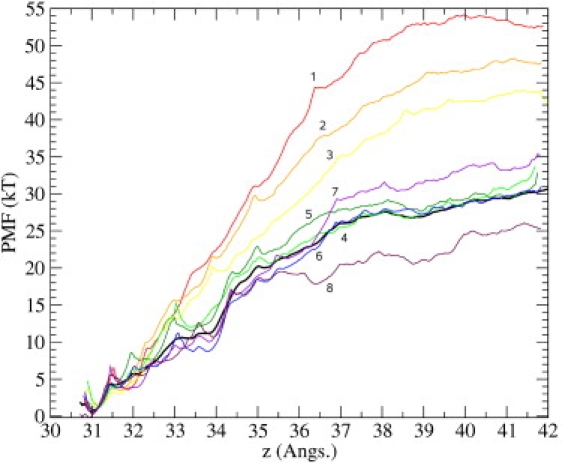
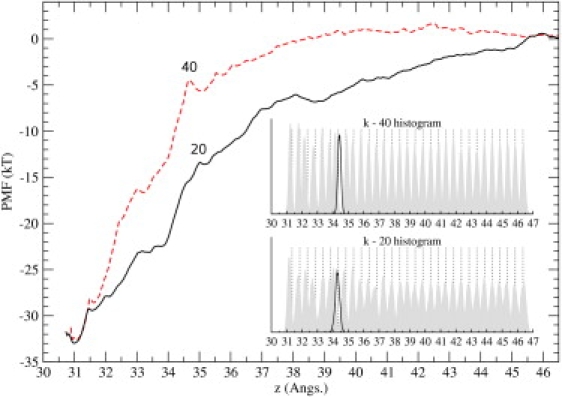
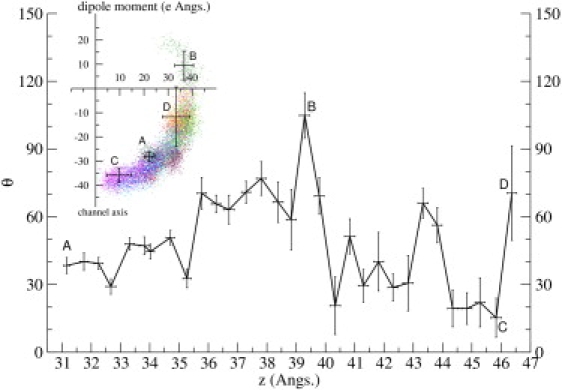
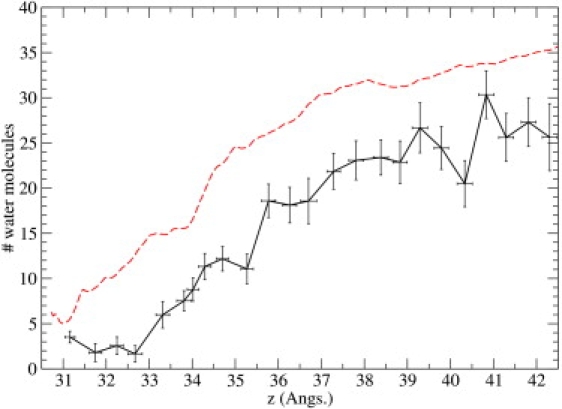
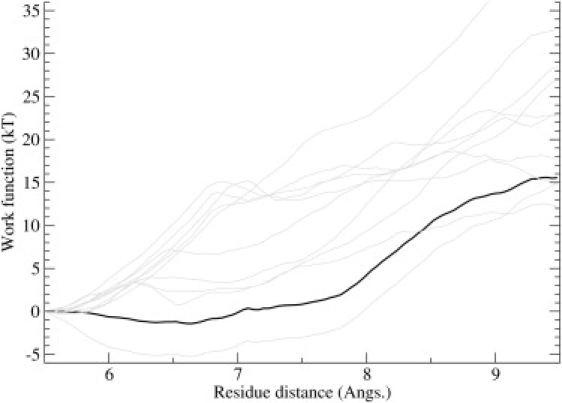
Ion channel-toxin complexes are ideal systems for computational studies of protein-ligand interactions, because, in most cases, the channel axis provides a natural reaction coordinate for unbinding of a ligand and a wealth of physiological data is available to check the computational results. We use a recently determined structure of a potassium channel-charybdotoxin complex in molecular dynamics simulations to investigate the mechanism and energetics of unbinding. Pairs of residues on the channel protein and charybdotoxin that are involved in the binding are identified, and their behavior is traced during umbrella-sampling simulations as charybdotoxin is moved away from the binding site. The potential of mean force for the unbinding of charybdotoxin is constructed from the umbrella sampling simulations using the weighted histogram analysis method, and barriers observed are correlated with specific breaking of interactions and influx of water molecules into the binding site. Charybdotoxin is found to undergo conformational changes as a result of the reaction coordinate choice--a nontrivial decision for larger ligands--which we explore in detail, and for which we propose solutions. Agreement between the calculated and the experimental binding energies is obtained once the energetic consequences of these conformational changes are included in the calculations.
Figures




References
-
- Fersht A. Freeman; New York: 1999. Structure and Mechanism in Protein Science.
-
- Halperin I., Ma B., Wolfson H., Nussinov R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins. 2002;47:409–443. - PubMed
-
- Brooijmans N., Kuntz I.D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003;32:335–373. - PubMed
-
- Gabdoulline R.R., Wade R.C. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 1998;14:329–341. - PubMed
-
- Wang W., Donini O., Reyes C.M., Kollman P.A. Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001;30:211–243. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
