

Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: immunopathogenesis and therapeutic implications
- PMID: 19349365
- PMCID: PMC2671245
- DOI: 10.2353/ajpath.2009.080987
Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: immunopathogenesis and therapeutic implications
Abstract
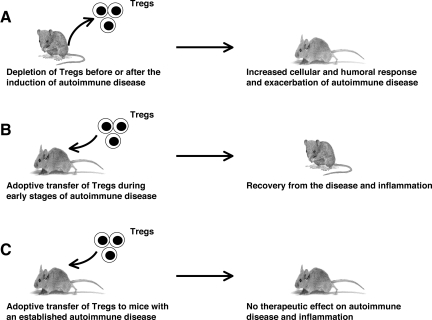
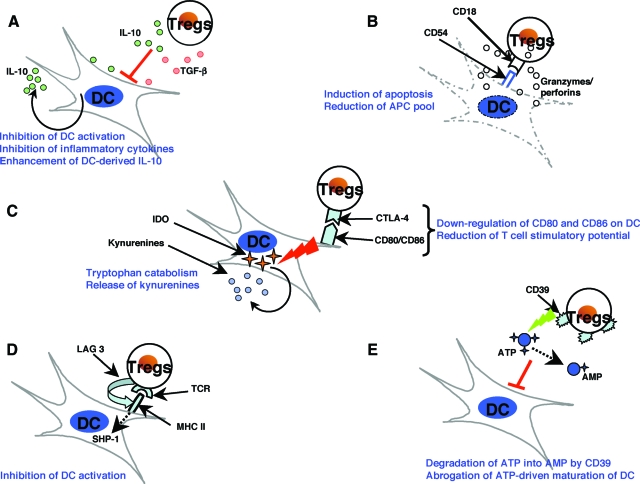
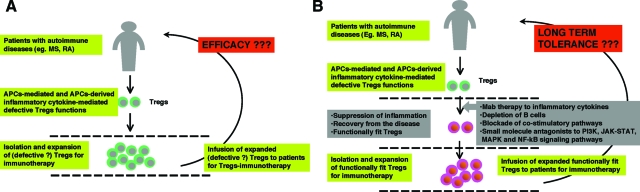
CD4+CD25+ regulatory T cells (Tregs) play a critical role in preventing immune aggression. One way in which Tregs exert immune surveillance activities is by modifying the function of antigen presenting cells (APCs) such as dendritic cells, macrophages, and B cells. Tregs can induce apoptosis of APCs or inhibit their activation and function, thereby regulating subsequent innate and adaptive immune responses. These actions of Tregs are mediated by both soluble factors (interleukin [IL]-10, transforming growth factor-beta, perforins, granzymes) and cell-associated molecules (cytotoxic T lymphocyte antigen 4, lymphocyte activation gene-3, CD18, neuropilin-1, LFA-1/CD11a, CD39), of which cytotoxic T lymphocyte antigen 4 has a key role. However, in autoimmunity, chronically activated APCs under the influence of intracellular signaling pathways, such as phosphatidyl inositol 3 kinase, JAK-STAT, MAPK, and nuclear factor-kappaB pathways, can escape surveillance by Tregs, leading to the activation of T cells that are refractory to suppression by Tregs. Moreover, APCs and APC-derived inflammatory cytokines such as tumor necrosis factor, IL-6, IL-1beta, and IL-23 can render Tregs defective and can also reciprocally enhance the activity of the IL-17-producing pathogenic Th17 T cell subset. Emerging knowledge of the importance of APC-Treg interactions in maintaining immune tolerance and aberrations in this cross talk in autoimmune diseases provides a rationale for therapeutic approaches specifically targeting this axis of the immune system.
Figures
References
-
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. - PubMed
-
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. - PubMed
-
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. - PubMed
-
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
